
希少難治性心筋症の病態をヒトiPS細胞由来分化心筋細胞により解明
研究成果のポイント
- 拡張型心筋症の原因は多様で、画一的な診断、治療方針では限界があるため、拡張型心筋症の克服には個々の重症例に対する新たなアプローチが必要であった。
- 重症心不全に至った拡張型心筋症症例に対して解析を行い、デスモグレイン2欠損が病因となることを同定した。さらに、疾患iPS細胞由来分化心筋細胞を用いて、ゲノム編集による遺伝子変異の修復、またはアデノ随伴ウイルスによる遺伝子の補充により、低下した収縮力が回復することを証明した。
- 遺伝子補充の治療概念を実証したことにより、希少難治性心筋症に対する新たな治療法の確立が期待される。
概要
大阪大学大学院医学系研究科の肥後修一朗特任准教授(常勤)(重症心不全内科治療学共同研究講座)、宮川繁特任教授(常勤)(最先端再生医療学)、彦惣俊吾准教授、坂田泰史教授(循環器内科学)らの研究グループは、希少難治性心筋症の病態をヒトiPS細胞由来分化心筋細胞により解明しました。
今回、研究グループは、若年で特発性拡張型心筋症と診断され重症心不全に至った症例に対して、遺伝子解析、心筋病理解析を行い、本症例の病因がデスモグレイン2欠損であることを見出しました。デスモグレイン2は細胞同士をつなぎ留め、収縮する力を伝達する介在板を構成するタンパク質で、これまでに、デスモグレイン2が完全に欠損することにより重症心不全に至った症例は世界でも報告はありませんでした。疾患iPS細胞を用いて、三次元心筋組織を作成することで、分化心筋における不整脈・心筋収縮力低下を再現しました。さらに、疾患iPS細胞に対してゲノム編集を用いて変異を修復したiPS細胞を構築し、同様の解析を行ったところ、不整脈・心筋収縮力が改善することを証明しました。また、アデノ随伴ウイルスによる遺伝子導入においても収縮力が改善することを明らかにしました。今後、希少難治性心筋症に対する新たな治療の可能性が期待されます。
本研究成果は、英国科学誌「Human Molecular Genetics」に、5月5日(水)に公開されました。
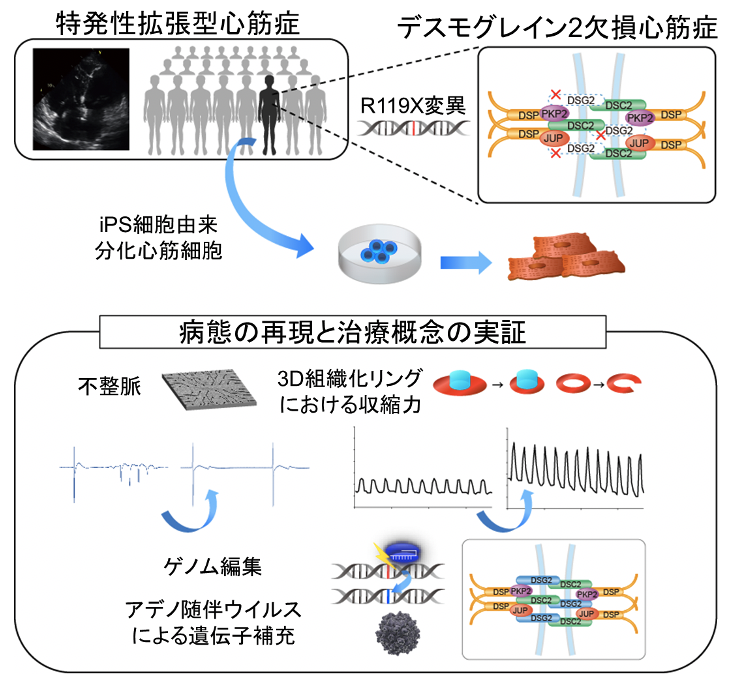
図1. デスモグレイン2欠損心筋症の同定
研究の背景
拡張型心筋症は、左心室収縮力の低下と左心室の拡大を生じる指定難病のひとつです。拡張型心筋症の原因は極めて多様であり、標準的な内科治療が奏功せず若年で重症化する症例が存在し、画一的な診断、治療方針は限界を迎えています。しかしながら、急速に重症心不全に至る拡張型心筋症の原因や病態メカニズムは依然明らかではなく、拡張型心筋症の克服のためには、個々の重症例に対する新たなアプローチが必要とされています。
本研究の成果
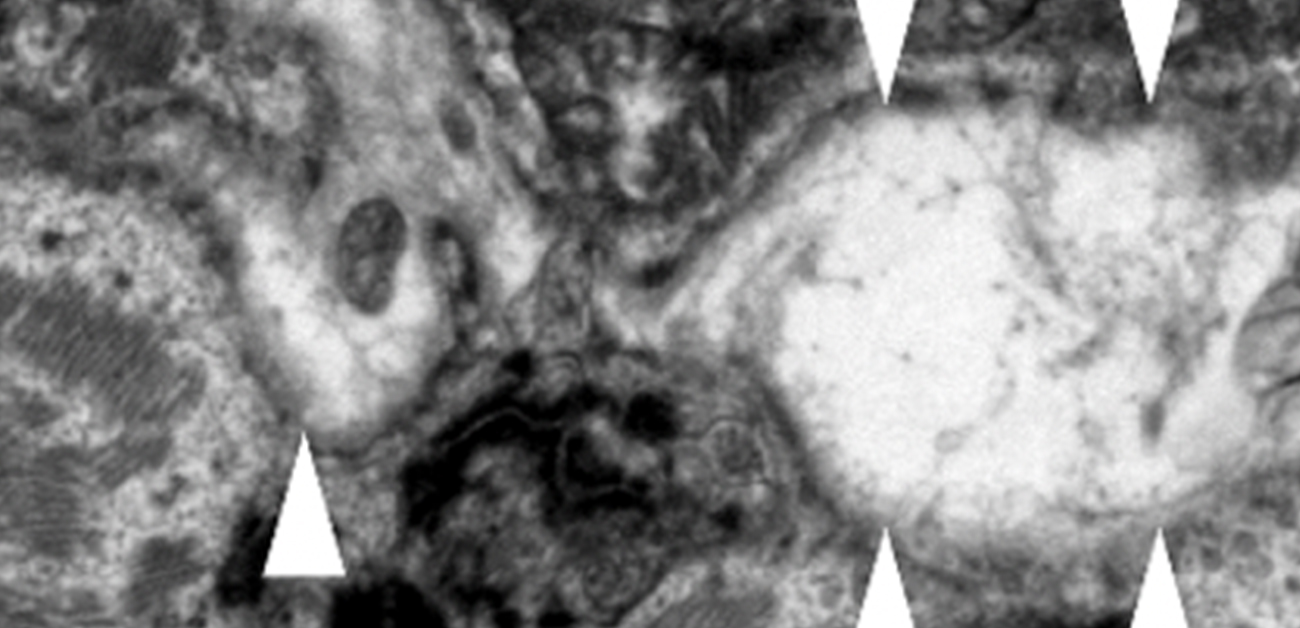
研究グループは、若年で原因不明の特発性拡張型心筋症と診断され、難治性心室性不整脈及び両心室拡大を呈した症例に遺伝解析を行い、介在板を構成するタンパク質であるデスモグレイン2(DSG2遺伝子)において、対立遺伝子の両方において正常なタンパク質が合成されないような変異を有する、ホモ接合型ナンセンス変異(c.C355T, p.R119X)を同定しました。デスモグレイン2は、心筋細胞の介在板に存在するタンパク質で、遺伝子変異により不整脈源性心筋症を発症することが知られていますが、ホモ接合型変異により完全欠損となる症例は、これまでに報告がありません。本症例の左室補助人工心臓植込み手術時に得られた左室心筋検体を用いて免疫染色を行ったところ、ホモ接合型R119X変異のため、心筋組織介在板におけるデスモグレイン2の発現は完全に欠損し、心筋細胞における空胞変性像や介在板構成タンパク質の空胞周囲への異常沈着像を認めました(図2左)。更に透過型電子顕微鏡を用いて微細構造を観察したところ、顕著な介在板の離開像を認め(図2右)、本症例は希少遺伝子変異により介在板構造が破壊されたデスモグレイン2欠損心筋症と考えられました。
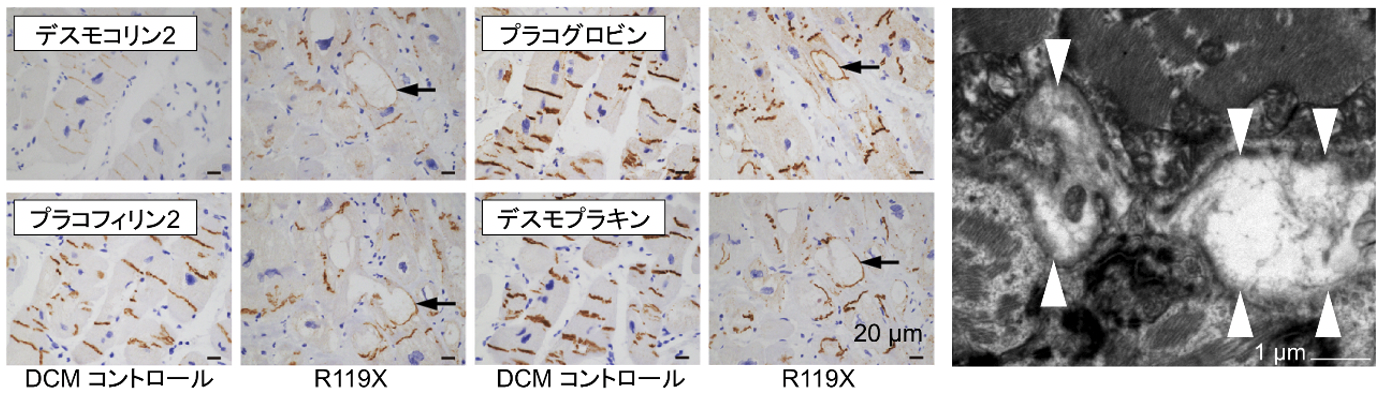
図2. (左)左室心筋組織における介在板構成タンパク質の異常局在・空胞内沈着
(右)左室心筋組織における介在板の離開像
デスモグレイン2欠損心筋症の病態メカニズムを解明するため、疾患iPS細胞(R119X-iPSC)を樹立しました。さらに、ゲノム編集技術を用いて片アレルの遺伝子変異を正常に修復し、デスモグレイン2発現を回復させたiPS細胞(HDR-iPSC)を作成しました。これらのiPS細胞を心筋細胞(CM)に分化させ、それぞれの機能を解析するため、微小電極アレイを用いた電位解析を行ったところ、R119X-iPSC-CMでは異常電位の出現(不整脈)を認めたのに対し、HDR-iPSC-CMでは認めませんでした(図3左)。さらに、自発性循環進行波刺激による三次元自己組織化リングを作成し、マイクロスケール圧縮強度測定装置を用いて、収縮力解析したところ、R119X-iPSC-CMでは顕著な組織構造の脆弱性及び微弱な収縮力が観察されましたが、いずれもHDR-iPSC-CMにおいては回復しました(図3右)。

図3.(左)微小電極アレイを用いた電位解析
(右)三次元組織化iPS分化心筋を用いた微小収縮力測定
iPS分化心筋の組織形成過程を経時的に観察したところ、R119X-iPSC-CMでは心筋組織構造形成が阻害され、心筋線維の菲薄化を認めました(図4左)。更に、透過型電子顕微鏡で観察したところ、介在板構造の離開所見が認められたのに対し、HDR-iPSC-CMではこれらの異常は認められませんでした。アデノ随伴ウイルスを用いてデスモグレイン2遺伝子をR119X-iPSC-CMに導入したところ、低下していた収縮力が回復しました(図4右)。
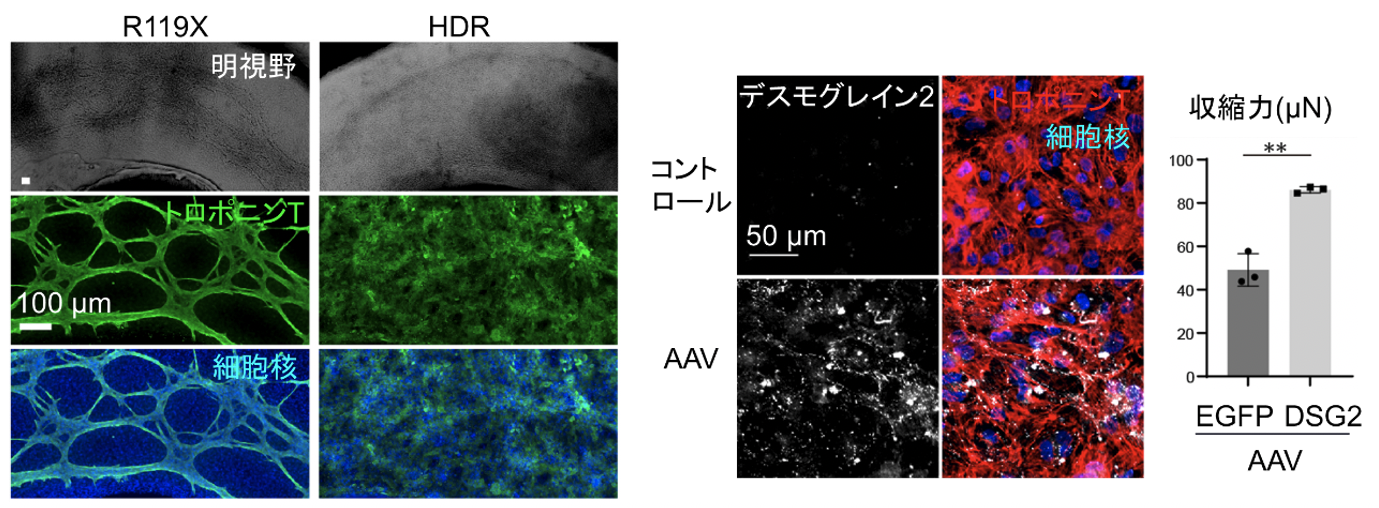
図4.(左)iPS分化心筋の組織形成過程
(右)R119X-iPS分化心筋へのアデノ随伴ウイルスによるデスモグレイン2遺伝子導入
本研究成果が社会に与える影響(本研究成果の意義)
2021年現在、日本では心臓移植希望登録者数は900名を超え、その多くを難治性心筋症が占めます。本研究成果は、特発性(原因不明)と診断される拡張型心筋症のなかに介在板構造が破綻したデスモグレイン2欠損心筋症が存在することを明らかとし、iPS分化心筋を用いてその病態と遺伝子補充の治療概念を実証しました。今後の難治性心筋症に対する精密医療の実現に寄与すると考えられます。
特記事項
本研究成果は、2021年5月5日(水)(米国東部時間)に英国科学誌「Human Molecular Genetics」(オンライン)に掲載されました。
【タイトル】 “Phenotypic Recapitulation and Correction of Desmoglein-2-deficient Cardiomyopathy using Human Induced Pluripotent Stem Cell-derived Cardiomyocytes”
【著者名】 Mikio Shiba1, Shuichiro Higo1,2, Takumi Kondo1, Junjun Li3,4, Li Liu3,4, Yoshihiko Ikeda5, Yasuaki Kohama1, Satoshi Kameda1, Tomoka Tabata1, Hiroyuki Inoue1, Satoki Nakamura2,6, Maki Takeda3, Emiko Ito3, Seiji Takashima6, Shigeru Miyagawa3, Yoshiki Sawa3, Shungo Hikoso1, Yasushi Sakata1
【所属】
1) 大阪大学大学院医学系研究科 循環器内科学
2) 大阪大学大学院医学系研究科 重症心不全内科治療学共同研究講座
3) 大阪大学大学院医学系研究科 心臓血管外科学
4) 大阪大学大学院医学系研究科 組織再生デザイン学
5) 国立循環器病研究センター 病理部
6) 大阪大学大学院生命機能研究科 医化学
なお、本研究は、国立研究開発法人日本医療研究開発機構再生医療実現拠点ネットワークプログラム(疾患特異的iPS細胞の利活用促進・難病研究加速プログラム)「難治性心筋症疾患特異的iPS細胞を用いた集学的創薬スクリーニングシステムの開発と実践」、日本学術振興会科学研究費助成事業の助成を得て行われました。
用語説明
- 拡張型心筋症
心臓の筋肉が収縮する力が低下し、心室(主に左心室)が大きく広がる病気で、難病に指定されている。
- デスモグレイン2
細胞-細胞間接着構造の一つであるデスモソームの構成タンパク質で、細胞接着分子。
- 介在板
細胞と細胞の間に存在する構造で、細胞どうしをつなぎとめ、収縮する力を伝達する役割がある。