
自己免疫疾患に関わるT細胞の制御分子を同定
免疫細胞の病原性獲得のしくみ
研究成果のポイント
・遺伝子発現の制御分子であるSatb1が、自己免疫疾患を引き起こすヘルパーT細胞の自分の体を攻撃する機能を制御する仕組みを明らかにした。
・Satb1はT細胞の分化に重要であることは分かっていたが、生体内でサイトカインを産生するヘルパーT細胞でのSatb1の役割についてはこれまで不明であった。
・今回明らかになった分子メカニズムを標的とすることで、自己免疫性ヘルパーT細胞が関わる自己免疫疾患の新しい免疫学的な治療法開発に結びつく。
概要
大阪大学の安田圭子医員(医学部附属病院、医学系研究科腎臓内科学)、坂口志文特任教授(常勤)(免疫学フロンティア研究センター)および京都大学の廣田圭司准教授(ウイルス・再生医科学研究所兼大阪大学招へい准教授)らの研究グループは、遺伝子発現の制御分子であるSatb1 に着目し、IL-17サイトカインを産生するヘルパーT細胞(Th17細胞)が病気を引き起こす仕組みを、ヒトの自己免疫疾患である多発性硬化症のマウスモデルを用いて明らかにしました。
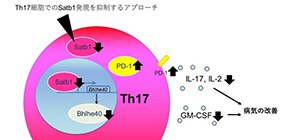
これまで、Th17細胞が病気を引き起こす機能を獲得するために、IL-17を産生するようになった後にどのような仕組みが働くのか分かっていませんでした。坂口特任教授らのグループは、T細胞の分化において色々な遺伝子の発現に作用する転写因子Satb1に着目し解析を行った結果、Th17細胞でのSatb1の欠損は、多発性硬化症マウスモデルにおいて、病気の発症に必須のサイトカインであるGM-CSF の産生を抑え、さらに免疫チェックポイント分子 の一つであるPD-1の発現を上昇させることで病気を軽減させる (図1) ことが分かりました。今後、Th17細胞でのSatb1の発現を抑えることを治療の標的とすることで、多発性硬化症のような自己免疫疾患に対する新しい治療法の開発に結び付くことが期待されます。
本研究成果は、2019年2月1日に英国科学誌「Nature Communications」(オンライン)に掲載されました。

図1 Th17細胞でのSatb1発現を抑えることで、炎症性サイトカイン(GMCSF, Il-17, IL-2)の産生が抑制され、自己免疫疾患に対する新規の治療となる可能性がある。
研究の背景
サイトカインの一種であるIL-17を産生するヘルパーT細胞(Th17細胞)には、1)体の正常な機能を保つ(腸管での生体恒常性の維持、真菌感染に対する防御)、2)自己免疫疾患(多発性硬化症、乾癬、関節リウマチ等)において自分の体を攻撃して病気をおこす、といった両方の働きがあります。Th17細胞が病気を引き起こす機能(病原性)を獲得するには、IL-17を産生するようになった後に、さらにIL-1βやIL-23といった炎症性のサイトカインの暴露を受けることが重要であることが、これまでに知られています (図2) 。しかしながら、生体内でIL-17を産生するようになった細胞を生きた状態で回収して性質を調べることが困難であったため、病原性Th17細胞を制御する詳細な仕組みについては明らかになっていませんでした。
これまでに、廣田准教授らの研究グループは、IL-17を産生したことがある細胞を蛍光色素で標識するリポーターマウスの作製に成功し、研究を進めていました。
今回、坂口特任教授らの研究グループは、T細胞の分化において色々な遺伝子の発現に作用する転写因子Satb1に着目し、Th17細胞がIL-17産生後、どのように病原性を獲得するかを調べました。
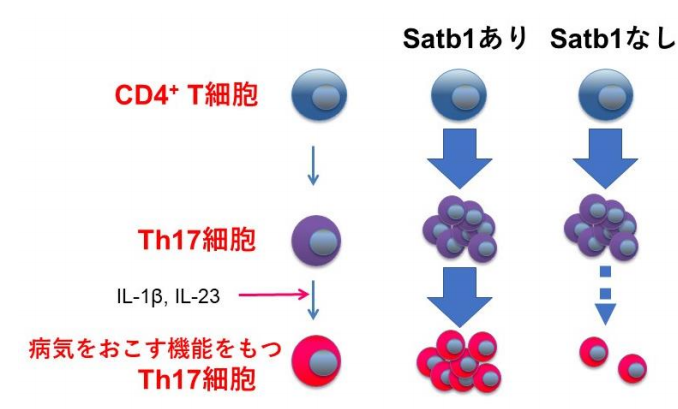
図2 Th17細胞の分化におけるSatb1の役割
本研究の成果
研究グループは、IL-17産生リポーターマウスを用いて、IL-17を産生後にSatb1の発現を欠損させ、同時にそれらの細胞を蛍光色素で標識するマウスを作製することで、Satb1を欠損するTh17細胞をマウス生体から(細胞が)生きた状態で回収することに成功しました。
まず、サイトカインを産生する前段階のT細胞である、CD4 + T細胞からIL-17の産生能を獲得する(Th17細胞に分化する)過程ではSatb1の有無による影響は見られず (図2) 、腸管に存在する病気をおこさないタイプのTh17細胞ではSatb1がなくても機能に影響しないことが分かりました。
次に、Th17細胞においてSatb1を欠損するマウスは、多発性硬化症マウスモデル(EAE:実験的自己免疫性脳脊髄炎)で、病気になりにくいことを見出しました (図3) 。
そのマウスの脳脊髄や炎症を起こしているリンパ節に含まれるTh17細胞について、サイトカインの産生能やタンパク質発現、遺伝子発現を解析したところ、Th17細胞でのSatb1の欠損は、EAEモデルにおいて、病気の発症に必須のサイトカインであるGM-CSFの産生を抑え、さらに免疫チェックポイント分子の一つであるPD-1の発現を上昇させることで病気を軽減させることが新たに分かりました (図4) 。
さらに、Satb1の発現を制御するサイトカインの種類を調べた結果、Th17細胞がIL-23に暴露されるとSatb1の発現が亢進し、逆にTGF-βではSatb1の発現は抑えられることが明らかになり、Th17細胞でのSatb1の発現は細胞をとりまく環境中に存在するサイトカインの影響を受けて変化し、さらにその結果としてTh17細胞が病気を引き起こすかどうかを決定することが分かりました。
Satb1がTh17細胞の機能を制御する下流の仕組みについて、さらに詳細に解析を行なった結果、Bhlhe40という転写因子の発現を調節する遺伝子領域にSatb1が転写因子として直接作用して、Bhlhe40の発現を亢進させること、さらにBhlhe40の作用によって、EAEにおいて病気をおこすサイトカインとして知られるGM-CSFの産生を増加させることが明らかとなりました。加えて、別の経路として、EAEの脳脊髄に浸潤しているTh17細胞において、Satb1の欠損は免疫チェックポイント分子の一つであるPD-1の発現を亢進する方向に働き、IL-17やIL-2といったサイトカインの産生を抑制し、結果としてEAEの病勢を抑えることが分かりました。

図3 Th17細胞においてSatb1を欠損すると、多発性硬化症モデルで病気になりにくい
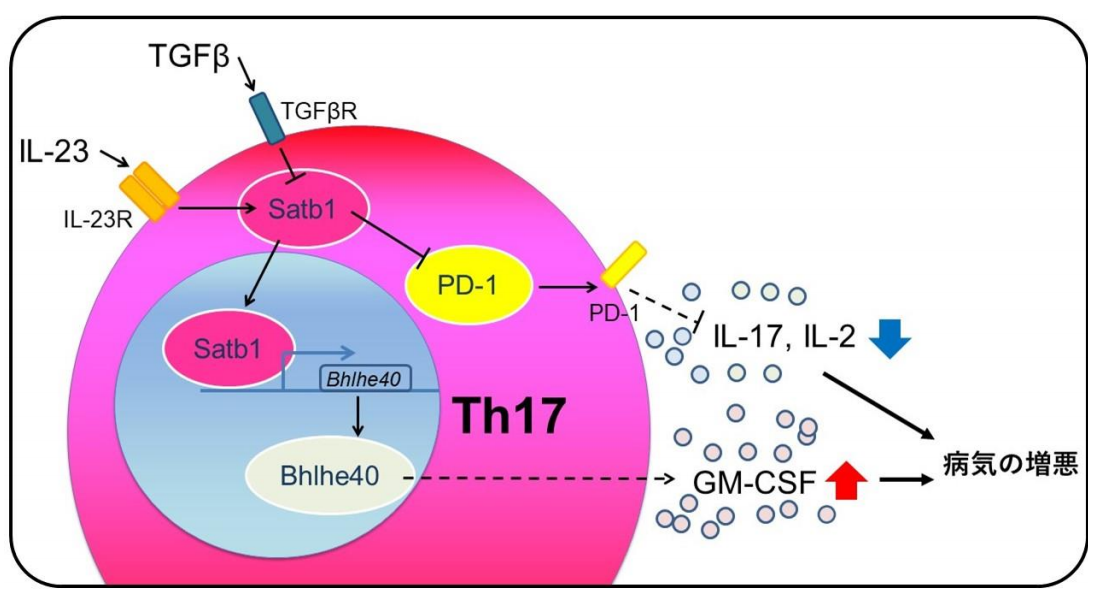
図4 Satb1による、病気を起こす機能を持つTh17細胞を制御するしくみ
本研究成果が社会に与える影響(本研究成果の意義)
本研究の結果から、Th17細胞でのSatb1の発現を抑えることを治療の標的とすることで、多発性硬化症のような自己免疫疾患に対する新しい治療法の開発に結び付くことが期待されます。
特記事項
本研究成果は、2019年2月1日に英国科学誌「Nature Communications」(オンライン)に掲載されました。
【タイトル】“Satb1 regulates the effector program of encephalitogenic tissue Th17 cells in chronic inflammation”
【著者名】Keiko Yasuda 1,2,3 , Yohko Kitagawa 2 , Ryoji Kawakami 2 , Yoshitaka Isaka 3 , Hitomi Watanabe 4 , Gen Kondoh 4 , Terumi Kohwi-Shigematsu 5 , Shimon Sakaguchi 1,2,* & Keiji Hirota 2,4,* .(*責任著者)
【所属】
1. 京都大学 ウイルス・再生医科学研究所 生体再建学分野
2. 大阪大学 免疫学フロンティア研究センター(IFReC) 実験免疫学
3. 大阪大学 大学院医学系研究科 腎臓内科学
4. 京都大学 ウイルス・再生医科学研究所 統合生体プロセス分野
5. カリフォルニア大学サンフランシスコ校 Department of Orofacial Sciences
本研究は、文部科学省科学研究費補助金「制御性T細胞による免疫応答制御の包括的研究(研究代表者:坂口志文)」、「炎症組織Th17細胞を起点とした炎症ネットワーク形成の分子基盤(研究代表者:廣田圭司)」、日本学術振興会特別研究員(安田圭子)の支援を受けて行われました。
また本研究は、京都大学ウイルス・再生医科学研究所およびカリフォルニア大学と共同で行われました。
参考URL
大阪大学 大学院医学系研究科 腎臓内科
http://www.med.osaka-u.ac.jp/pub/kid/kid/index.html
用語説明
- Satb1
(サットビーワン、special AT-rich sequence-binding protein-1)/Terumi Kohwi-Shigematsu教授らのグループにより1994年に最初に同定された。核内に存在するタンパク質であり、転写因子として他の遺伝子の発現を調節する働きをもつことが分かっているが、その際に一挙に多くの遺伝子の発現を調節できることが既に明らかになっている。T細胞の分化において、どの種類のT細胞になるかを決定する遺伝子を直接的に調節することが分かっており、制御性T細胞が正常に分化するためにはSatb1が必須であることを報告した坂口志文特任教授(常勤)らの研究グループを含め、複数のグループからそれぞれの種類のT細胞での重要性が報告されている。
- GM-CSF
(Granulocyte Macrophage colony-stimulating Factor:顆粒球マクロファージコロニー刺激因子)/骨髄の多能性造血幹細胞に働き、白血球の分化を促すサイトカインの一種。造血性のサイトカインとしての働き以外に、自己免疫疾患の発症および増悪に関わるサイトカインであることが明らかになってきている。
- 免疫チェックポイント分子
T細胞が働く際には、抗原提示細胞に対して抗原特異的なナイーブT細胞が結合するだけでなく、さらに補助刺激分子を介した補助シグナルがないと、サイトカインを産生し、ヘルパーT細胞としての働きを示さない仕組みになっている。抑制性補助刺激受容体の中には、PD-1やCTLA-4といった、ヘルパーT細胞としての働きを抑制する方向に働く免疫チェックポイント分子が含まれている。例えば、がん細胞の一部では、このPD-1の発現を上昇させることで、T細胞からの排除を免れているものがあり、この仕組みを解除する免疫チェックポイント阻害剤はがんに対する新規の治療法として開発され、既に臨床応用されている。