
When Good Cells Go Bad: Regulating the MS-Causing Properties of Th17 Cells
Researchers identify a protein regulator responsible for inducing the pathogenic properties of Th17 cells, leading to multiple sclerosis and other inflammatory autoimmune disorders
Inflammatory and autoimmune diseases such as psoriasis, rheumatoid arthritis, and multiple sclerosis (MS) are a major healthcare burden worldwide and are life-altering conditions for afflicted patients. Yet, while much is known about the mechanisms of disease, in most cases there are very few effective treatment options.
However, a recent study published in Nature Communications provides hope for autoimmune disease sufferers. Led by researchers from Osaka University, the study team found that genome organizer protein Satb1 triggers a shift in immune cells, inducing tissue inflammation and autoimmunity. This breakthrough could lead to new treatments targeting the source of inflammation.
Interleukin 17-producing T-helper (Th17) cells are a recently-identified class of immune cells that play a major role in protecting us against invading pathogens. But, despite their name, Th17 cells aren’t always so helpful. In fact, studies have shown that excessive inflammation induced by Th17 cells is actually one of the underlying causes of many inflammatory and autoimmune disorders.
But what causes Th17 cells to cross the line from helpful to harmful?
To answer this question, the researchers examined genome organizer protein Satb1. “We have known for some time that Satb1 is indispensable for the development of T-cells in the thymus. However, how it is involved in the regulation of pathogenic processes of Th17 cells in inflamed tissues had not been examined,” explains lead author Keiko Yasuda.
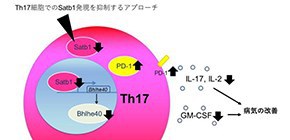
Using a mouse model of MS, the researchers deleted the Satb1 gene in Th17 cells and found that the disease no longer developed in the mutant mice. Key to this mechanism was the lack of expression of a protein called granulocyte-macrophage colony-stimulating factor (GM-CSF) by the Satb1-defective Th17 cells. GM-CSF is a pathogenic cytokine that causes localized tissue inflammation in MS and other inflammatory autoimmune diseases. Importantly, Satb1 was also found to enhance the expression of the GM-CSF gene, confirming its role in promoting inflammation in diseased individuals.
But as well as promoting inflammation, Satb1 also seems to block the expression of a protein specially designed to suppress T-cell inflammatory activity and prevent autoimmune diseases. This protein, programmed cell death protein 1, was more highly expressed in Satb1-deficient Th17 cells compared with normal Th17 cells, indicating that Satb1 normally inhibits protein expression.
As explained by study corresponding author Shimon Sakaguchi, these findings are something to get excited about: “Our results suggest that manipulating Satb1 gene expression in Th17 cells could form the basis of novel treatments for various autoimmune diseases caused by Th17 cells. If we can prevent the pathogenic processes of Th17 cells, we may be able to alleviate or even eliminate disease symptoms.

Figure 1 . The role of Satb1 in Th17-cell differentiatioSatb1 is dispensable for the differentiation of Th17 cells but plays a pivotal role in the effector functions of pathogenic Th17 cells. (credit: Osaka University)

Figure 2. Satb1 controls the pathogenic function of Th17 cells via regulation of Bhlhe40 and PD-1 expression. Satb1 is indispensable for the effector functions of pathogenic Th17 cells, including GM-CSF and IL-17 production via regulation of Bhlhe40 and PD-1 expression, respectively, in a murine autoimmune disease model. (credit: Osaka University)
This research was conducted in cooperation with the Institute for Frontier Life and Medical Sciences, Kyoto University and University of California.
The article “Satb1 regulates the effector program of encephalitogenic tissue Th17 cells in chronic inflammation” is published in Nature Communications at DOI: https://doi.org/10.1038/s41467-019-08404-w .
Related links