
非結核性抗酸菌症に対する迅速・正確な病原体同定手法を開発
175種を網羅した大規模ゲノムデータベース構築
研究成果のポイント
・これまで詳細な同定が困難であった非結核性抗酸菌(NTM;Non-Tuberculous Mycobacteria )の正確かつ迅速な同定手法を新たに開発した。
・NTMがひきおこすNTM症の治療方針決定のためには,およそ200種が知られるNTMに対して亜種レベル の正確な同定が要求されるため、今回、大規模ゲノムシーケンスによるデータベースの拡張とゲノム配列の比較法を組み合わせることで、NTM175種に対する少量のデータによる亜種同定を実現した。
・すでに実際のNTM症患者から単離された29検体についても亜種レベルでの同定に成功しており、今後のNTMの迅速な亜種同定・早期診断による治療・予防方法の確立が期待される。
概要
大阪大学微生物病研究所松本悠希特任研究員(常勤)と中村昇太特任准教授(常勤)らの研究グループは、琉球大学大学院医学研究科の金城武士助教らと共同で、少量のデータで非結核性抗酸菌(NTM;Non-TuberculousMycobacteria)の菌種を迅速・正確に見分ける(同定する)手法を開発しました。
NTMとはマイコバクテリウム属細菌 のうち結核菌群やらい菌を除いた種の総称であり、人の肺や皮膚にNTM症 を引き起こす病原体として知られています。菌種・亜種ごとに治療方針が異なるため、適切な治療を行うためには亜種レベルでの正確な同定が必要となりますが、多種多様なNTMの亜種を正確に同定できる方法はこれまで存在していませんでした。
今回、中村特任准教授らの研究グループは、大規模なゲノムシーケンス (図1) によってNTMのゲノムデータベースの拡張、さらにMLST(Multi-Locus Sequence Typing) と呼ばれる、ゲノム配列を比較して同定を行う手法とあわせて用いることで、正確 (図2A) かつ迅速 (図2B) にNTMを同定する手法を開発しました。今回の研究成果により、10分程度のシーケンスで得られるごく少量のデータから、175種にも及ぶNTMの正確な同定が可能となりました。また、NTM症患者から分離されたNTMについて、従来の手法では同定できなかった16株を含む29株すべてを亜種レベルまで同定することに成功しました。
本研究成果により、NTMが正確に同定されることで、NTM症患者は病原体に応じた適切な治療を受けられるようになることが期待されるとともに、同定にかかる時間が劇的に短縮される可能性があります。これにより、NTM症の早期発見による予防や新たな治療方法の確立へ貢献することが期待されます。
本研究成果は、英国科学誌「Emerging Microbes & Infections」に、2019年7月9日(火)に公開されました。

図1 NTM同定のワークフロー。
MinIONシーケンサー(図中央)を用いてゲノム配列を解読し、同定を行う。
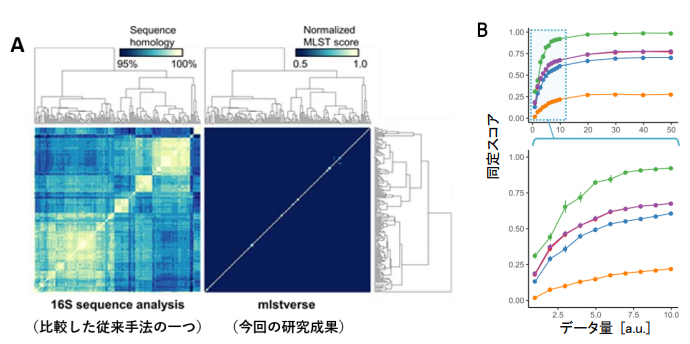
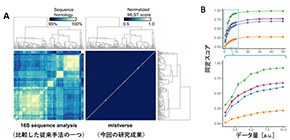
図2
A)従来手法(左)と今回開発した同定法(右)の比較。白い点が相同性の高いスコア、つまりターゲットとなる菌種である可能性が高いことを表す。従来法では複数の種に対して高いスコアを示したのに対し、今回開発した手法ではターゲットとなる種に対してのみ高いスコアを示した。
B)データ量と同定スコア。今回開発した手法ではX軸の値が低い位置でも同定スコアがプラトーに達しており、極めて少ないデータ量で菌種を同定できていることがわかる。
図は論文中より抜粋。
研究の背景・内容
マイコバクテリウム属細菌はさまざまな感染症を引き起こすことが知られており、大きく分けて結核を引き起こす結核菌、ハンセン病の原因となるらい菌、そしてそれら以外の非結核性抗酸菌(NTM;Non-Tuberculous Mycobacteria)の3つに分類されます。NTMは主に呼吸器感染症を引き起こしますが(肺NTM症)、本邦を含め世界的にその罹患率の増加が報告されています。結核菌、ハンセン病については予防法や治療法が確立されているのに対して、NTM症にはいずれの対策も十分に確立されていないことから、公衆衛生上、重要な感染症に位置付けられています。NTM症の治療方針を立てる上でまず重要なことは原因菌種・亜種の同定ですが、従来法では亜種レベルまでの正確な同定はできないのが問題となっていました。
中村特任准教授らの研究グループでは、複数の遺伝子の塩基配列を同定に用いるMLSTをマイコバクテリウム属細菌の同定に応用することで、極めて高速・高精度な同定を可能にする手法を新たに開発しました。またそれを実現するために、ゲノム情報が十分に知られていなかった63種のNTMについて、最新の次世代シーケンシング技術 を用いることで、全ての菌種においてほぼ完全長のゲノム配列を得ることに成功しています。得られたゲノム配列は同定のためのデータベース構築に利用されたほか、それらを比較解析することにより、歴史的経緯から過去にM.terraeとして統合されていたM.novumがゲノム配列の観点から別種である可能性などが新たに示唆されました。
本研究成果が社会に与える影響(本研究成果の意義)
本研究成果により、NTMが正確に同定されることで、NTM症患者は病原体に応じた適切な治療を受けられるようになることが期待されます。肺NTM症の中でも特に難治性で、近年本邦で増加しているM.abscessusは、日常診療レベルではこれまで亜種レベルの同定ができなかった種の一つですが今回の成果により、同定が可能となりました。
また本研究成果を応用することにより同定にかかる時間が劇的に短縮される可能性があります。現在一般的に行われているNTM同定では、まず細菌を特別な培地で培養することで単離してくる必要がありますが、NTMは生育の極めて遅いものもおり、培養には数か月を要することもあります。本研究成果は少量のデータからでも菌種同定が可能であるため、NTM症患者の喀痰などに含まれるわずかなゲノムDNAから、培養なしに直接NTMの同定を成し得る可能性があります。これによりNTM症の早期発見による予防や新たな治療方法の確立へ貢献することが期待されます。
研究者のコメント
NTMは我々の非常に身近な水場や土壌にも生息しているため、感染のリスクはどうしても避けられません。NTMはとても強固な細胞壁を持っているため、抗生物質に強い耐性を示すものが多く、一度症状が進行してしまうと、完治まで非常に長い時間がかかることが知られています。本研究の成果の応用がNTM症の早期発見に繋がり、患者の皆様の治療に生かされることを期待します。
特記事項
本研究成果は、2019年7月9日(火)に英国科学誌「Emerging Microbes & Infections」(オンライン)に掲載されました。
タイトル:“Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547genomic profiles”
著者名:Yuki Matsumoto, Takeshi Kinjo, Daisuke Motooka, Daijiro Nabeya, Nicolas Jung, Kohei Uechi, Toshihiro Horii, Tetsuya Iida, Jiro Fujita, Shota Nakamura
なお、本研究は、沖縄感染症事業,革新的先端研究開発支援事業(AMED-CREST)、戦略的情報通信研究開発推進事業(SCOPE)ならびに科学研究費助成事業の一環として行われ、琉球大学大学院医学研究科藤田次郎教授との共同研究として行われました。
参考URL
大阪大学 微生物研究所 病原体同定研究グループ 中村研究室
http://nkmr.biken.osaka-u.ac.jp/
用語説明
- 非結核性抗酸菌
NTM:
マイコバクテリウム属細菌はグラム陽性細菌に分類される真正細菌の一属で、結核菌など約200種が登録されています。このマイコバクテリウム属の細菌は抗酸菌と総称され、そのうち結核菌群および、らい菌を除いた細菌を非結核性抗酸菌(NTM)といいます。これらにより引き起こされる感染症はNTM症と呼ばれ、免疫不全患者だけでなく健常者へも感染し、感染後は自覚的な症状がほとんど無いまま長い時間をかけて病状が進行します。発症後は咳・痰・血痰・発熱・食欲不振・体重減少・全身倦怠感などが見られ、抗生物質も効きづらいため、長期の適切な薬剤治療が必要となる厄介な病気です。
- 亜種レベル
生物の分類区分で、種の下位区分。非結核性抗酸菌症の主要な病原菌であるMycobacterium aviumの亜種であるhominissuis、silvaticum、paratuberculosisなど、近年次々と亜種が発見されている。
- MLST
Multi-Locus Sequence Typing:
データベースに登録された配列と同定したい検体の配列を照合することによりマッチする菌種を探す手法はsequencetypingと呼ばれ、それを複数遺伝子に拡張したものがMLSTと呼ばれます。菌種固有の遺伝子のDNA配列をデータベースに登録しておくことで極めて高い精度で同定が行え、データベースの規模が大きいほど検出能力が上昇します。今回、本研究においてはリボソーム分子の構成に関わる遺伝子やNTMの抗生物質耐性に関わると考えられている184遺伝子を選んだ上で、公共データベースおよび我々が新規に解読した計175種のNTMのゲノム情報を用いることで、マイコバクテリウム同定のためのデータベースを完全に新規に作成しました。
- 次世代シーケンシング技術
全ての生物が持つゲノムDNAはA(アデニン)、T(チミン)、G(グアニン)、C(シトシン)の4種類の塩基が連なった構造をしています。DNAの塩基配列を解読することはシーケンシングと呼ばれ、近年の技術革新が可能とした多量の塩基配列の同時解読技術を、特に次世代シーケンシング技術と呼びます。本研究においてはIllumina社のHiSeq2500とOxford Nanopore Technologies社のMinIONという性質の異なる複数の次世代シーケンサーを組み合わせることで、より高精度なゲノムの構築を実現しました。