
がんゲノム情報×細胞シミュレーションで個別化医療へ大きく前進
誰にでも使える創薬支援解析ツールを開発・公開!
研究成果のポイント
・遺伝子発現情報のみから実験をせずに細胞中の薬剤標的分子の活性を定量的に予測する計算手法の開発
・計算手法の高度化によるがんゲノムデータを用いた細胞シミュレーション解析ツールの構築・公開
・個人の遺伝子特性を反映した患者固有モデルの構築を実現することで創薬の加速や個別化医療の進展に期待
概要
大阪大学蛋白質研究所細胞システム研究室(岡田眞里子教授)の研究グループは、同大学大学院理学研究科の大学院生井元宏明さん(博士後期課程)を中心に、個別医療を目標とした患者固有モデルの構築に向けた計算手法を開発し、それに基づき、がんゲノム情報と細胞シミュレーション技術を組み合わせた創薬支援解析ツールを構築・公開しました。
これまでがん治療においては、がんの大きさ、浸潤の程度、疾患マーカーの有無などのいくつかの指標に基づき治療法が選択されてきました。しかし、これまでの分類法では患者の予後に大きな違いが生じることから、個々の患者の遺伝子情報に基づいた新たな分類法の開発が急務となっています。本研究では、がんの層別化(たくさんのがんゲノムデータを遺伝子の特徴や薬剤応答によってグループ分けすること)や個別化医療を目標とした患者固有モデル の構築を目的としました。
そのために、公共データベース から取得したがん細胞株のRNAシーケンス(網羅的な遺伝子発現)データと細胞シミュレーション を組み合わせ、がん細胞増殖の標的であり、薬剤探索の指標となるキナーゼ(リン酸化酵素)の活性を定量的に予測する計算手法を開発しました。
この方法を用いると、複数の細胞株の実験データを数理モデルに学習させることにより、RNAシーケンスデータのみを入力として、学習していない別の細胞株のキナーゼの活性値を実験を行わずにシミュレーションのみで予測できるようになるため、創薬研究現場の大幅な効率向上が可能となります。
さらに、この手法をもとに、細胞実験データの種類や量を増やし、数理モデルや解析法の精緻化と拡張を行っていけば、細胞株データを用いて学習させた数理モデルに患者検体由来のRNAシーケンスデータを入力することにより、患者固有モデルの構築が可能になると考えられます。また、そのことにより、がんの層別化や患者ごとに適した薬剤の探索が可能になると考えられます。
本研究では、多くのがんで重要な働きを担うErbB(HER)受容体シグナル伝達系のネットワーク(313パラメータ、39遺伝子)について、分子間の結合解離・酵素反応・分子局在・分解などを多数の文献をもとに常微分方程式により再現した数理モデルを構築し、乳がん細胞株を対象に研究を進めました。数理モデルは、ネットワークを構成する分子、分子の量などの初期値、分子間の結合強度などの動的なパラメータから構成されており、これらの要素、量、パラメータを変えることにより、薬剤探索や薬剤評価に資するシミュレーションが可能となります。本解析では、感度解析 という手法により、多くのがんにおいて薬剤耐性時の活性化上昇が認められ薬剤標的としても研究が進められるAktキナーゼ(プロテインキナーゼ B) の分子活性を指標に、Akt活性の制御に適した分子および適さない分子を同定しました。また、本解析からは細胞ごとの遺伝子発現量の差異が、遺伝子変異などよりも、それぞれの細胞内での分子の実活性や細胞の性質を決める上で、重要な指標となり得る可能性が示唆されました。
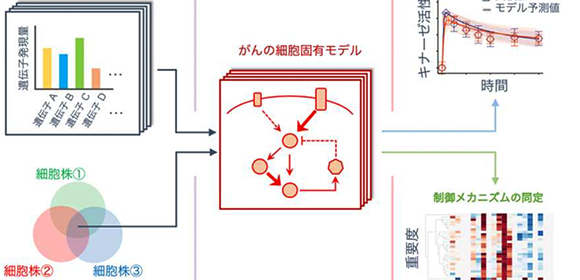
このような数理モデルを用いた細胞シミュレーション技術は、疾患メカニズムの同定や定量的解析において優れており、ビッグデータに基づく分類に優れた人工知能(AI)と相補的に創薬研究に用いられることが期待されています。本研究における細胞シミュレーションの解析基盤は、学習実験データをもとにモデルの動的パラメータを最適化するパラメータ最適化法 、遺伝子ネットワークの重要な要素を同定する感度解析、公共データベースから遺伝子発現情報を取得するツール、シミュレーション・解析結果を簡単に可視化する解析ツールを含み、BioMASS(Modeling and Analysis of Signaling Systems)は、パッケージ化し、オープンソースソフトウェアとして公開しています( https://github.com/okadalabipr/biomass ) (図1) 。この解析ツールは、細胞モデリングに必要な一連の技術を集積し、モデリングに馴染みのない人でも約一週間で解析ができるようになります。
本研究成果は、国際科学誌『Cancers』に、10月7日(水)(日本時間)に公開されました。

図1 公共ゲノムデータを用いた細胞シミュレーションによる細胞固有モデルの構築と分子活性および制御分子の予測。細胞のリン酸化実験データをもとに学習させた数理モデルに公共データベースから取得したRNAシーケンスデータを入力(インプット)することにより、創薬標的であるキナーゼの実活性値を定量的に予測し、その活性制御に関わる重要遺伝子を感度解析により同定(アウトプット)。
研究の背景および内容
生命科学分野では、細胞や組織の遺伝子発現や変異情報などのさまざまなシーケンスデータが公共データベースに蓄積され、基礎・応用研究に利用されるようになりました。特に創薬研究においては、コスト、スピード、倫理面からも、このようなデータの利活用が重要視されています。このようなデータ解析の手法としては、統計や機械学習などの手法が広く知られていますが、これらの手法は基本的に莫大な数の均一なデータを必要とし、相関などに基づいた薬剤や遺伝子の分類には適しますが、創薬開発に必要となる分子メカニズムなどの因果関係の予測は不得手なことから、新たな計算手法の開発が必要でした。
また、がん研究においては、従来のサブタイプ分類 を超えた患者の遺伝子情報に基づく層別化や個別化治療へのニーズが近年高まっています。しかし、研究現場では培養細胞やモデル動物を用いた細胞や組織内の分子活性を指標とする実験主導の研究が中心で、ヒト研究への橋渡しのためには、多くのコストとステップを必要としました。また、患者由来の細胞の培養には高度な技術が必要とされ、多くの研究室で日常的に実験することは困難でした。そのため、遺伝子情報を用い、コンピュータ上でひとりひとりの患者をバーチャルに再構成する"患者固有モデル(patient-specific model)"に関する研究が世界中で進められるようになりました。
しかし、遺伝子情報はあくまでもそのデータが取得された細胞や組織の一瞬の状態を捉えたもので、細胞などが置かれた生体環境における動的な振る舞いを反映していません。よって、このような患者固有モデル構築のためには、遺伝子情報に加え、患者の細胞本来の振る舞いを再現するための動的なパラメータが必要となります。そして、このパラメータの取得を行うためのパラメータ最適化には、本来、患者組織そのものから取得した細胞を培養し、多様な生育環境下で実験データを取得することが必要であり、この労力および技術的な困難さが患者固有モデルの開発のボトルネックとなっていました。また、取得実験値に対して、遺伝子のネットワークが大きいこと、実験値に多くのノイズが含まれることなどの生物学研究の特性もパラメータ最適化の問題となっていました。さらに、このようなモデルの構築や解析のためには、プログラミング技術やアルゴリズムなどの高度な知識を必要とし、生物学や医学に関わる実験研究者が日常的に携わることはこれまで困難でした。
本研究では、患者固有モデルの構築を最終目的として、公共データベースから取得可能ながん細胞株のRNAシーケンスのデータを用い、細胞固有モデルを構築することを目的としました。そして、複数の乳がん培養細胞株から取得した3種類の分子(キナーゼや転写因子)のリン酸化修飾量の時系列変化の実験データに対して、39遺伝子から成る数理モデルを用い、動的パラメータの学習を行うことにより、学習していない別の乳がん細胞株の分子リン酸化の実活性値をRNAシーケンスデータのみから予測する計算手法を開発しました。この手法を精査し、拡張していけば、培養細胞株から学習させた動的パラメータに患者固有のRNAシーケンスデータを入力することにより、患者固有モデルの構築が可能になると考えられます。
さらに、本研究では、人工知能(AI)の一種である遺伝的アルゴリズム(GA; genetic algorithm)を用い、モデルの学習データ(培養細胞の実験値)に対してパラメータ最適化計算を一定時間、並行して計算し、パラメータセットを多数取得する戦略を取っています。このことにより、限られた実験データに対して多数のパラメータセットを計算的に取得することが可能になります。ある細胞の学習データに対して取得したパラメータセット群を、他の別の細胞へ適用することにより、RNAシーケンスデータのみから当該細胞内分子の実活性値を予測することができるようになりました。工学研究などで利用される本来のパラメータ最適化計算法は、レンズの焦点距離などといった局所の単一解を求めることを目的としますが、本研究では、生物の有する不確かさやノイズを考慮し、あえて単一解を求めず、生物の持つ性質として多数の解を得、それを分子活性予測に利用した点に、手法の新しさがあります。
また、本研究で開発した細胞シミュレーション解析ツールは、細胞モデリングに必要なモデルの記述、パラメータ最適化、重要因子を同定する感度解析などの一連の手法を集積し (図2) 、実験研究者が使いやすいように工夫を凝らしており、モデリングに馴染みのない人でもおおよそ一週間で解析ができるようになります。このツールは、生命科学研究だけでなく、工学など他分野のシミュレーション解析にも応用することができます。

図2 BioMASSの解析ツール。BioMASSでは(1)数理モデルの構築、(2)動的パラメータの最適化、(3)重要分子を同定する感度解析をプログラミングや数値計算の深い知識がなくともシームレスに行うことができる。
本研究成果が社会に与える影響(本研究成果の意義)
本研究により、公共データベースなどから患者の臨床検体から取得したRNAシーケンスデータが得られれば、実験をせずとも、直接、細胞シミュレーションにより、バーチャルに分子標的薬の効果を評価できる可能性が示されました。がん領域ではシグナル伝達系薬剤を対象とした分子標的薬が多く開発され、その有効性の高さから、現在も活発に開発が進められています。これまでの創薬研究では、樹立培養細胞や患者から取得したオルガノイドを用いた実験解析が主体でしたが、樹立培養細胞では病態を正しく反映しておらず、オルガノイドに関しては高度な技術を必要とすることが課題でした。今回開発した計算手法並びに細胞シミュレーションとヒトの疾患ゲノム情報を組み合わせた手法を、遺伝子変異やエピゲノム情報などを含め、高度に発展することができれば、実験を行わなくとも、さまざまな公共データを組み合わせて研究者の目的に合わせた解析が可能となります。特にシーケンスデータに関しては、1検体のシーケンス費用は、現在6万円前後と高額であることから、1000人のRNAシーケンスデータを公共利用することにより、約6000万円の直接経費に加え、臨床試料の取得とそのための人件費、時間、スペースなどの巨額な総額研究費用を削減することができます。また、細胞の数理モデルそのものもデータベースに多数登録されており、本解析ツールを用いて、すでに構築された数理モデルを目的に合わせて組み合わせ解析することも可能となります。本研究の発展は、創薬研究のスピードアップ、コストの削減、患者ごとに適した薬剤を選択する個別化医療において大きなブレークスルーとなることが期待されます。
特記事項
本研究成果は、2020年10月7日(水)(日本時間)に国際科学誌『Cancers』(オンライン)に掲載されました。
論文タイトル: "A computational framework for prediction and analysis of cancer signaling dynamics from RNA sequencing data—Application to the ErbB Receptor Signaling Pathway"
著者: Hiroaki Imoto, Suxiang Zhang, Mariko Okada
掲載雑誌: Cancers (Basel)
なお、本研究は、科学技術振興機構(JST)未来社会創造事業探索加速型「共通基盤」領域の研究開発課題「創薬を加速する細胞モデリング基盤の構築(研究開発代表者:岡田眞里子)」および日本学術振興会科学研究費補助金基盤研究(A)「疾病機序理解のための遺伝子ネットワーク数理モデル基盤の構築」などの支援を得て行われました。
参考URL
蛋白質研究所細胞システム研究室HP
http://www.protein.osaka-u.ac.jp/cell_systems/index_ja.html
用語説明
- 細胞シミュレーション
細胞シミュレーションに関する研究は、ヒトの全ゲノム解読前後の2000年頃から本格的にはじまり、アメリカ、ドイツ、日本で活発に進められてきました。細胞の中の遺伝子間の相互作用や活性を数理モデルで記述し、コンピュータ上で再現する手法で、細胞の制御原理の理解や創薬に利用されています。細胞シミュレーションに用いられる数理モデルには、統計や物理法則など目的に応じて異なる原理を用いますが、本研究では、生化学反応の表現に用いられる連立微分方程式モデルを用いています。遺伝子の量や相互作用を情報として含むことから、疾患ゲノム解析により入手可能な遺伝子発現量や変異の情報などを入力値としてシミュレーションすることが可能になります。このことにより、患者固有の細胞シミュレーションが可能になると考えられています。
- 患者固有モデル
これまでのがん研究により、さまざまながんはひとつまたはそれ以上の遺伝子の変異の組み合わせにより起こること、またそのことは患者ごとに異なることが明らかになりました。そのため、これまでのような平均化した患者像でなく、コンピュータ上でひとりひとりの患者を、遺伝子情報をもとにバーチャルに再構成する"患者固有モデル(patient-specific model)"が求められるようになりました。このような研究は海外で活発に進められており、患者の層別化や薬剤のオーダーメード化に貢献すると考えられています。
- 公共データベース
生命科学研究では、培養細胞、モデル動物、ヒト疾患における遺伝子やたんぱく質の網羅的計測値は、データベースに保存されており、公共利用が可能です。例えば、米国NIHの運営するThe Cancer Genome Atlas(TCGA)プログラムでは、乳がんだけに限っても約1000人の患者のゲノムおよび予後の情報が入手可能です。本研究ではCancer Cell Line Encyclopedia(CCLE)という培養細胞株のデータベースの乳がん細胞由来の遺伝子発現情報を利用し、解析に用いました。また、これまでに発表された数理モデルはBioModelsという公共データベースに登録されており、本基盤を用いて、研究目的に合わせてモデルの解析と統合が可能となります。
- 感度解析
感度解析は、遺伝子発現量やパラメータの変化などの摂動を与えた場合の細胞ネットワーク出力に対する影響度(感度)を調べる手法です。本研究では、Akt活性を出力とした場合に、その活性化に影響力の大きい遺伝子を特定しました。
- Aktキナーゼ(プロテインキナーゼ B)
Aktは、シグナル伝達系に属するセリン・スレオニンキナーゼのひとつで、成長因子、サイトカイン、インスリンなどの細胞への投与により活性化され、代謝、細胞増殖、細胞死などを引き起こす重要な分子です。Aktは他のシグナル伝達系の分子を活性化したり抑制したりする役割がありますが、多様ながんにおいて、シグナル伝達系阻害剤への耐性が見られる場合に、活性が上昇することが報告されています。そのため、Aktをコントロールできれば、薬剤耐性をコントロールできると考えられ、Aktを標的とした薬剤開発が進められています。
- パラメータ最適化
細胞シミュレーションのためのモデル構築には、遺伝子間の相互作用の強さ、物質の合成分解速度、局在などの動きに関するパラメータが必要となります。しかしながら、細胞中の速度定数を直接測定することは困難なため、細胞シミュレーションでは、パラメータ最適化手法を用いた予測を行うことがほとんどです。本研究では、細胞におけるネットワークの遺伝子の発現量の情報とたんぱく質のリン酸化の計測値を入力として、それを再現する速度定数を遺伝的アルゴリズム(GA)により推定しています。遺伝的アルゴリズムは、人工知能(AI)に使用される進化計算の一種で、本研究では、たんぱく質のリン酸化データを説明することのできるパラメータ候補をランダムに選択、交叉、変異を与えながら、正しい解を選んでいきます。
- サブタイプ分類
がんのサブタイプ分類:
乳がんの場合、サブタイプは分子マーカーの発現の有無により4つに分類されます。具体的には、エストロゲン受容体(ER)、ErbB2(HER2)受容体のどちらか一つが発現有、2つ共に発現有、2つ共に発現無という分類です。ERにはER拮抗剤など、ErbB2には抗体などの代表的薬剤があり、サブタイプに基づいた薬剤治療が現在行われています。しかし、最近のゲノム解析により、この4つのサブタイプ群それぞれにおいても、個々の患者の遺伝子の変異や発現の多様性が高く、効果的な薬剤の選択や治療のためには、新たな患者の再分類(層別化)が必要だと考えられています。