
大規模ゲノムの機械学習手法により日本人集団の地域による多様性を解明
日本人のゲノムを知り、ゲノム個別化医療に役立てる
研究成果のポイント
・日本人集団17万人のゲノム配列に最新の機械学習手法を適用し、日本の中でも地域による細かなゲノムの多様性が存在することを、視覚的に分かりやすく示した。
・この機械学習手法をイギリス・アラブ・マレーシアのゲノムデータにも適用し、世界の国々の中の地域性を反映した詳細なゲノムの多様性を明らかにした。
・日本人集団内部の多様性は、ゲノム情報による将来の病気のリスク予測にも無視できない影響を与えており、個別化ゲノム医療の社会実装には多様性への深い理解が重要であることが示唆された。
概要
大阪大学大学院医学系研究科遺伝統計学教室坂上沙央里大学院生(東京大学大学院医学系研究科博士課程)、岡田随象教授(理化学研究所生命医科学研究センター客員主管研究員)らの研究グループは、日本・イギリス・アラブ・マレーシアのゲノムデータに機械学習 を応用し、これまで見つけられなかった地域ごとの詳細なゲノムの多様性を視覚的に明らかにする手法を発表しました。更に、将来の疾患罹患リスクをゲノムにより予測する手法であるポリジェニック・リスク・スコア(PRS) に、この多様性が思いがけない影響を与える可能性を示しました。
私達現生人類は、アフリカで誕生した共通の祖先から、ヨーロッパ、中東、アジアへと移住し、その過程でさまざまな環境に適応し多様性を増してきました。その多様性は現在の人々のゲノム配列に反映されており、古典的な線形次元削減法 である主成分分析(PCA) の手法を用いると、ゲノム情報から大まかに世界の人種を特定できることが知られていました。しかし、例えば一つの国の中など、これまでゲノムからは同じ人種と分類されてきた人々の中にも各地域を特徴づけるような細かなゲノムの多様性が存在するかどうかについては未解明でした。
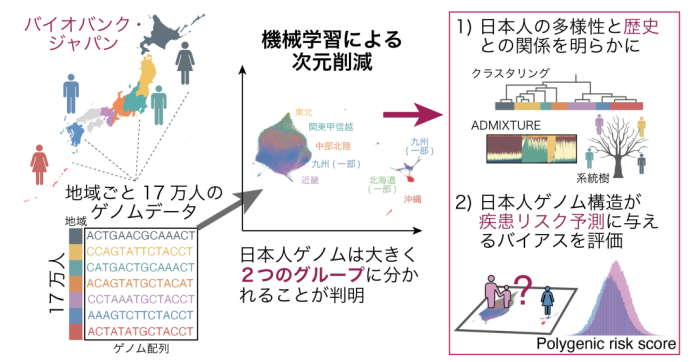
図1 日本人集団17万人のゲノムデータに機械学習・次元削減手法を適用し、詳細なゲノム多様性を明らかに
今回、岡田教授らの研究グループは、日本のさまざまな地域から集められた17万人規模のゲノムデータに対して、最近開発された機械学習・非線形次元削減 の手法を適用することで、地域ごとのゲノムの多様性を二次元座標に視覚的に分かりやすく描出する手法を提案しました (図1) 。この手法により、日本人集団はおおきく本州を中心とするグループと琉球を中心とするグループの大きく2つに分類されることが分かり、琉球グループは機械学習手法を再度適用すると更に詳細な分類が可能であることが判明しました。この機械学習手法をイギリス・アラブ・マレーシアのゲノムデータにも適用し、一つの人種集団内の多様性を明らかにしました。最後に、日本人集団内部のグループ構造は、ゲノム配列全域に分布する無数の遺伝的変異の情報を用いた疾患リスク予測値である「ポリジェニック・リスク・スコア」の値にも影響を与えることを示し、ポリジェニック・リスク・スコアに基づくリスク層別化には、一つの人種集団内部の緻密な多様性まで考慮した方法論が求められることを示しました (図1) 。
本研究成果は、米国科学誌「 Nature Communications 」に、3月26日(木)19時(日本時間)に公開されました。
研究の背景
私達一人ひとりが持つゲノム配列は、遥か昔の共通祖先から徐々に変化しながら受け継がれてきました。現代人のゲノム配列の個人差は、アフリカで数百万年前に誕生した人類が大陸を超えて移住し、集団の分化と統合を繰り返し環境に適応してきた結果、形成されました。近年ゲノム配列を解読する技術が飛躍的な進歩を遂げ、大規模なゲノムデータを比較して解析した結果、例えば寒冷地では脂肪を蓄えるように(Fumagalli et al. Science 2015)、標高が高い地域では酸素を効率的に得られるように(Huerta-Sánchez et al. Nature 2014)ゲノム配列が変化してきたことが分かりました。更に、古典的な線形次元削減法である主成分分析(PCA)の手法を用いると、ゲノム情報から大まかに世界の人種を特定できることも知られていました。しかし日本のように、多くの島々で構成され多様な文化を有する一つの国の内部にも、各地域を特徴づけるような細かなゲノムの多様性が存在するのかどうかについては未解明でした。その理由として、ヒトゲノム上の個人の遺伝的多型 は数千万箇所にも及び、地域による特徴をこの中から適切に選び出し、人間が見てわかるように視覚化することが困難であることが挙げられます。
本研究の成果
研究グループは、近年シングル・セル解析など機能ゲノム科学(functional genomics)の分野で応用されてきた機械学習・次元削減の手法である t-SNE 、UMAP をバイオバンク・ジャパン の日本人集団17万人のゲノム配列データに応用し、日本人集団が大きく2つに分類されることを明らかにしました (図2・上左) 。これは、先行研究(Yamaguchi et al. Am J Hum Genet 2008)で示された本州を中心とするグループと琉球を中心とするグループに相当しました。更に、琉球グループに対してもう一段階の次元削減を行うと、地理的に隣り合って位置していても異なる遺伝的背景を有するサブグループへと詳細な分類が可能であることが判明しました (図2・上右) 。
これらのグループが歴史的に形成された意義を確かめるために、系統樹解析などの集団遺伝学的手法を集約的に応用し、この大きな2つのグループ構造は、数万年前に東南アジアから移住してきた集団と数千年前に朝鮮半島から移住してきた集団との由来の違いに起因する可能性が考えられました。
今回提案した機械学習手法が日本だけでなく世界の人種集団の内部構造を明らかにすることができるか検証するために、イギリスのUKバイオバンク 、マレーシアとアラブのゲノムコホートとの共同研究を行いました。その結果、主成分分析(PCA)とUMAPという二つの機械学習の手法の組み合わせ(PCA-UMAP)が詳細な集団内のゲノム構造を解明することに長けていることが分かりました (図2・下) 。
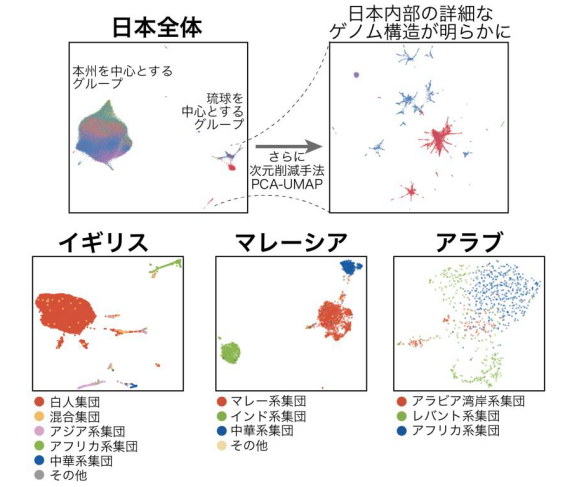
図2 非線形機械学習の手法であるPCA-UMAPをゲノムデータに適用し可視化した結果
(上)日本人集団17万人のゲノム配列にPCA-UMAPを適用して見つかった大きな2つのグループのうち、琉球グループに再度PCA-UMAPを適用。詳細なゲノム構造を明らかにした。(下)イギリス・マレーシア・アラブのゲノムデータにも機械学習・次元削減手法PCA-UMAPを適用し、ゲノム多様性を視覚化。
ゲノム配列による集団の多様性は、過去の私達の適応進化の歴史に示唆を与えるとともに、ゲノム情報を利用した将来の精密医療を適切に実現する上で重要な意味を持ちます。中でも、ポリジェニック・リスク・スコアはゲノム配列から将来の健康リスクや検査値を予測することができ、臨床現場でのゲノム情報によるリスクの層別化に応用されることが期待されています。しかし、このポリジェニック・リスク・スコアは人種によるゲノム構造の違いによって値の分布が異なるため、人種を超えた比較には注意を要することが知られていました(Martin et al. Nat Genet 2019)。研究チームは、今回、機械学習の手法により特定された詳細な日本人集団内部のゲノム構造もポリジェニック・リスク・スコアの分布に影響を与えるかどうかを、22種の疾患・45種の量的疾患に対して検証実験を行いました。その結果、本州中心のグループと琉球中心のグループには多くの形質でスコアの分布に違いが認められ、リスク予測のバイアスとなり得ることが強く示唆されました。
特に、身長とbody mass index(BMI; 肥満度の指標)ではその違いが顕著であり、ゲノム情報からの身長の予測値は実際の身長と同様に琉球を中心とするグループの方が小さかったのに対し、ゲノム情報からのBMIの予測値は琉球を中心とするグループの方が小さい一方で、実際のBMI値は琉球を中心とするグループの方が大きいことが判明しました (図3・上) 。この逆転現象を理解するため、過去50年のBMIの時系列データを解析したところ、沖縄を中心とした食生活の欧米化に起因する急速な肥満が原因となっている可能性が考えられました。すなわち、琉球グループにおいて本州グループを超えた肥満を認めるようになったのはこの10数年のことであり、ゲノム情報からのBMIの予測値が琉球グループにおいて小さかったのは、現在生きる世代の数世代前のBMIの予測値を反映しているためではないかと考えられました (図3・下) 。
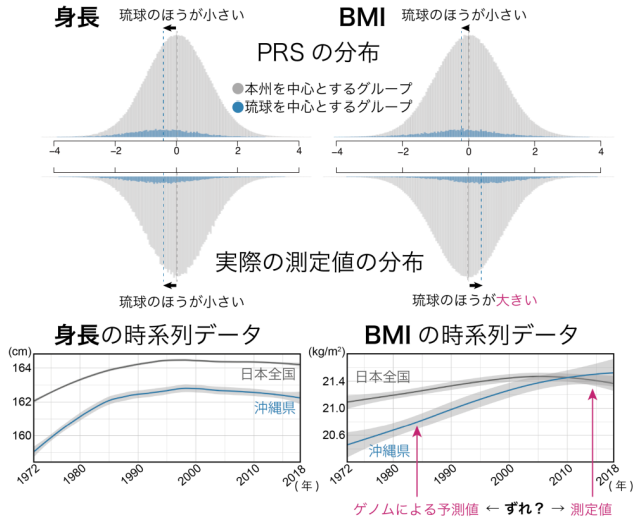
図3 次元削減手法により定義した2つのグループによる身長・BMIのPRSと検査値自体の分布と、過去50年間の身長・BMIの平均値の時系列変化
(上)身長とBMIのPRSは2つのグループにより分布が異なり、BMIは実際のデータと逆の動きを示した。(下)過去50年の時系列データより、身長は常に琉球グループの方が小さい一方で、ゲノムによる予測時期のBMIは琉球グループの方が小さく、実際の測定時期のBMIは琉球グループの方が大きい逆転現象が起きている。
本研究成果が社会に与える影響(本研究成果の意義)
今回の研究で、一つの国の内部にも存在するような詳細なゲノムの多様性が、思いがけないバイアスをポリジェニック・リスク・スコアに与えていること、そしてその理解には各地域の適応進化や歴史も理解する必要があることが分かりました。これは、将来の個別化ゲノム医療を実装していく上で重要な示唆を与えるものと考えられます。
特記事項
本研究成果は、2020年3月26日(木)19時(日本時間)に米国科学誌「 Nature Communications 」(オンライン)に掲載されました。
【タイトル】“Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction.”
【著者名】Saori Sakaue 1-3 ,Jun Hirata 1,4 , Masahiro Kanai 1,2,5 , Ken Suzuki 1 , Masato Akiyama 2,6 , Chun Lai Too 7,8 , Thurayya Arayssi 9 , Mohammed Hammoudeh 10 , Samar Al Emadi 10 , Basel K. Masri 11 , Hussein Halabi 12 , Humeira Badsha 13 , Imad W. Uthman14, Richa Saxena 15,16 , Leonid Padyukov 8 , Makoto Hirata 17 , Koichi Matsuda 18 , Yoshinori Murakami 19 , Yoichiro Kamatani 2,20 , Yukinori Okada 1,21,22
【所属】
1.大阪大学大学院医学系研究科 遺伝統計学
2.理化学研究所 生命医科学研究センター 統計解析研究チーム(研究当時)
3.東京大学大学院医学系研究科 アレルギー・リウマチ学
4.帝人ファーマ株式会社 創薬探索研究所
5.ハーバード大学医学部 Department of Biomedical Informatics
6.九州大学医学部 眼科
7.マレーシア保健省 Allergy and Immunology Research Center
8.カロリンスカ大学病院 Division of Rheumatology
9.コーネル大学カタール Department of Internal Medicine
10.ハマド医療法人 Department of Internal Medicine
11.ヨルダン病院 Department of Internal Medicine
12.キング・ファイサル専門病院 Rheumatology Division
13.エミレーツ病院 Dr. Humeira Badsha Medical Center
14.ベイルートアメリカン病院 Department of Rheumatology
15.ハーバード大学医学部麻酔科 Center for Genomic Medicine
16.ブロード研究所 Program in Medical and Population Genetics
17.東京大学医科学研究所 ヒトゲノム解析センター シークエンス技術開発分野
18.東京大学大学院新領域創成科学研究科メディカル情報生命専攻 クリニカルシークエンス分野
19.東京大学医科学研究所 癌・細胞増殖部門 人癌病因遺伝子分野
20.東京大学大学院新領域創成科学研究科メディカル情報生命専攻 複雑形質ゲノム解析分野
21.大阪大学免疫学フロンティア研究センター 免疫統計学
22.大阪大学先導的学際研究機構 生命医科学融合フロンティア研究部門
本研究は、日本医療研究開発機構(AMED)オーダーメイド医療の実現プログラム、ゲノム医療実現推進プラットフォーム事業先端ゲノム研究開発「遺伝統計学に基づく日本人集団のゲノム個別化医療の実装」の一環として行われ、大阪大学大学院医学系研究科バイオインフォマティクスイニシアティブの協力を得て行われました。本研究で使用したサンプルは、「オーダーメイド医療の実現プログラム」において収集されたものです。
研究者のコメント<大学院生 坂上沙央里>
私達一人ひとりが持つゲノム配列は、自分の現在の設計図であるとともに、遠い祖先から受け継がれてきた過去の歴史の記録、そして未来の健康リスクを予測する道具でもあります。アフリカで誕生し、わずか数百万年の間に驚くべき広範囲に移住し生活を営むようになった現生人類。新たな次元削減手法が明らかにした日本・世界の人々の間に存在する地域による幅広いゲノム多様性は、それぞれの地域の人々が多様な歴史をたどってきたことを意味します。この多様性の構造を理解し適切に反映することがゲノム医療の社会実装にも不可欠であることを今回の研究で再確認できました。
参考URL
大阪大学大学院医学系研究科 遺伝統計学HP
http://www.sg.med.osaka-u.ac.jp/index.html
用語説明
- 機械学習
統計モデル手法の一つであり、計算機を使用してデータを学習し、そのパターンを推測することで任意の課題を効率的に実行するためのアルゴリズムのこと。今回の研究では、次元削減、すなわち各個人の数千万箇所に及ぶゲノム多型データ(数千万次元のデータ)を、なるべく情報量を保ちつつ2次元に効率的に削減し、平面に視覚化する課題を実行するための機械学習を行った。
- ポリジェニック・リスク・スコア
(polygenic risk score; PRS):
大規模ゲノムワイド関連解析研究(GWAS; ヒトゲノム配列上に存在する数千万カ所の遺伝子変異とヒト疾患との発症の関係を網羅的に検討する、遺伝統計解析手法)により、疾患や形質との関連が示唆された数十〜数千の遺伝的変異の重み付きの和を個人ごとに計算したスコア。このスコアは実際の疾患発症リスクと相関することが示されており、集団内でスコアの分布を調べることで、特にその疾患のリスクが高い個人を特定することができる。
- 線形次元削減法
データの特徴を保ちながら次元を削減する方法のうち、線形変換を行う方法のこと。主成分分析 、多次元尺度構成法(MDS)、正準相関分析(CCA)などが挙げられる。
- 主成分分析
(Principal Component Analysis; PCA):
古典的な次元削減手法の一つであり、多次元のデータを分散が大きい順の直行成分に分解する手法。観測値はこれらの成分の線型結合として表すことができる。
- 非線形次元削減
データの特徴を保ちながら次元を削減する方法のうち、線形変換以外の変換を行う方法のこと。アイソマップ、カーネル主成分分析、t-SNE 、UMAP などが挙げられる。
- 遺伝的多型
ゲノム配列の個体差のうち、集団中に一定以上の頻度存在するもののこと。中でもゲノム塩基配列中に一塩基が変異した多様性である一塩基多型(single nucleotide polymorphism; SNP)が代表的である。
- t-SNE
(t-distributed Stochastic Neighbor Embedding):
機械学習による非線形次元削減手法の一つ。確率分布を利用して、高次元空間でのデータ点同士の類似度と低次元空間に削減した後の各データ点同士の類似度のKullback-Leiblerダイバージェンスを最小にするように変換する手法。
- UMAP
(Uniform Manifold Approximation and Projection):
最も新しい機械学習による非線形次元削減手法の一つ。リーマン幾何学と代数トポロジーに基づき、高次元空間のデータ構造を保ち、トポロジー間のクロス・エントロピーを最小にしながら低次元のデータに変換する手法。計算速度が t-SNEより高速であるのも特徴。
- バイオバンク・ジャパン
(BioBank Japan)/日本人集団27万人を対象にした生体試料のバイオバンクであり、ゲノム解析が終了した人数は約20万人とアジア最大である。オーダーメイド医療の実現プログラムを通じて実施され、ゲノムDNAや血清サンプルを臨床情報と共に収集し、研究者へのデータ提供や分譲を行っている。
- UKバイオバンク
(UK Biobank):
英国で実施されている国家的バイオバンク機構。中高年のボランティア約50万人を対象に、ゲノム情報や2000以上の多彩な臨床情報、追跡情報を収集し、ほぼ無償で世界の研究者にデータの公開や分譲を行っている。