
Redefining human diseases through the lens of your DNA
Researchers from Osaka University address inequities in genomic research by analyzing data for over 200 health-related traits and diseases in an Asian population
Throughout most of medical history, physicians have diagnosed individuals with various diseases based upon their description and presentation of clinical symptoms. In recent years, investigations using genome-wide association studies (GWAS) have helped scientists understand genetic factors involved in different diseases. In a newly published article in Nature Genetics, a team led by researchers at Osaka University and Harvard Medical School performed GWAS in biobank samples from various populations to identify specific genomic loci related to a diverse array of medical indications and traits.
Although GWAS have intensively been performed for the past decades, their widespread applicability has not been sufficient. Most GWAS have used European population data, with a relatively small number of phenotypes, as individuals with certain diseases were recruited for participation. A methodical way to interpret the results and put them in context has also been lacking. Thus, the Osaka University group aimed to address these limitations and perform equitable GWAS.
To achieve this, the team performed GWAS using BioBank Japan, which includes medical data from 180,000 people and is one of the largest non-European biobanks. This incorporated 220 health-related diseases and traits and made the study extremely diverse and comprehensive, particularly in Asian populations.
“We wanted to significantly expand the scope of this GWAS to gain as much meaningful insight from this biobank as possible,” says lead author of the study Saori Sakaue. “In fact, 108 of the phenotypes had never been part of GWAS in East Asian people,” says the co-first author of the study Masahiro Kanai.
The researchers then performed cross-population meta-analyses with the UK Biobank (United Kingdom) and FinnGen (Finland), which involved 628,000 individuals. This work helped identify over 14,000 genomic loci of phenotypic significance. Of these, 5,000 loci were novel discoveries. The group ensured their summary statistics were publicly available on a web portal (https://pheweb.jp) as a resource for geneticists and researchers worldwide.
“We believed it was extremely important to create a way to let others in the greater genetics community access our data,” explains Yukinori Okada, senior author. “We definitely want to foster global collaborations and hope meaningful functional follow-up experiments will be performed from our findings.”
Because of the complex nature of their GWAS results and disease genetics in general, the team performed statistical deconvolution of their summary statistics and other data. This step was important because it allowed the researchers to make disease-relevant conclusions from their enormous dataset.
“The deconvolution allowed us to pinpoint specific genetic variants and shared mechanisms associated with various diseases across populations. Those mechanisms were in turn useful in reevaluating and restructuring human diseases through the lens of human genetics,” explains Sakaue.
This work provides groundbreaking results that address pertinent predispositions within previous GWAS. The team’s findings will allow investigators to examine human diseases through genetics with an unbiased eye.
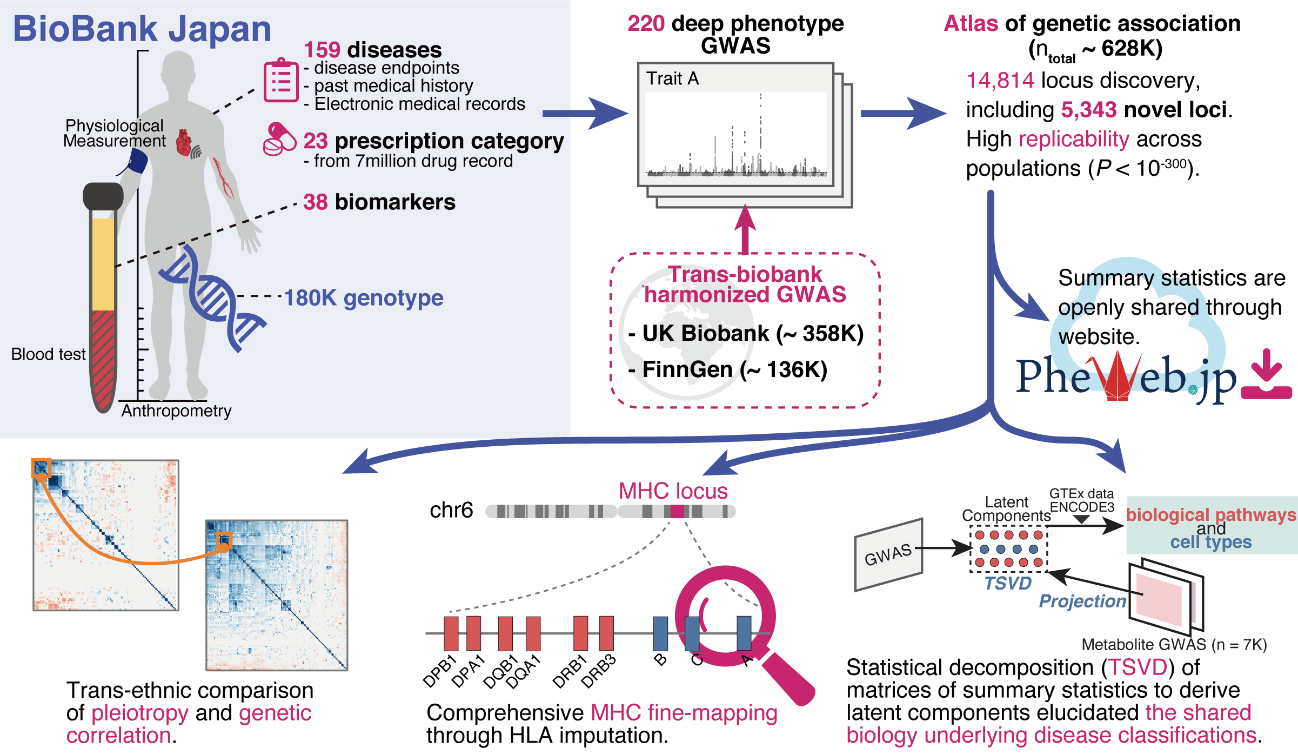
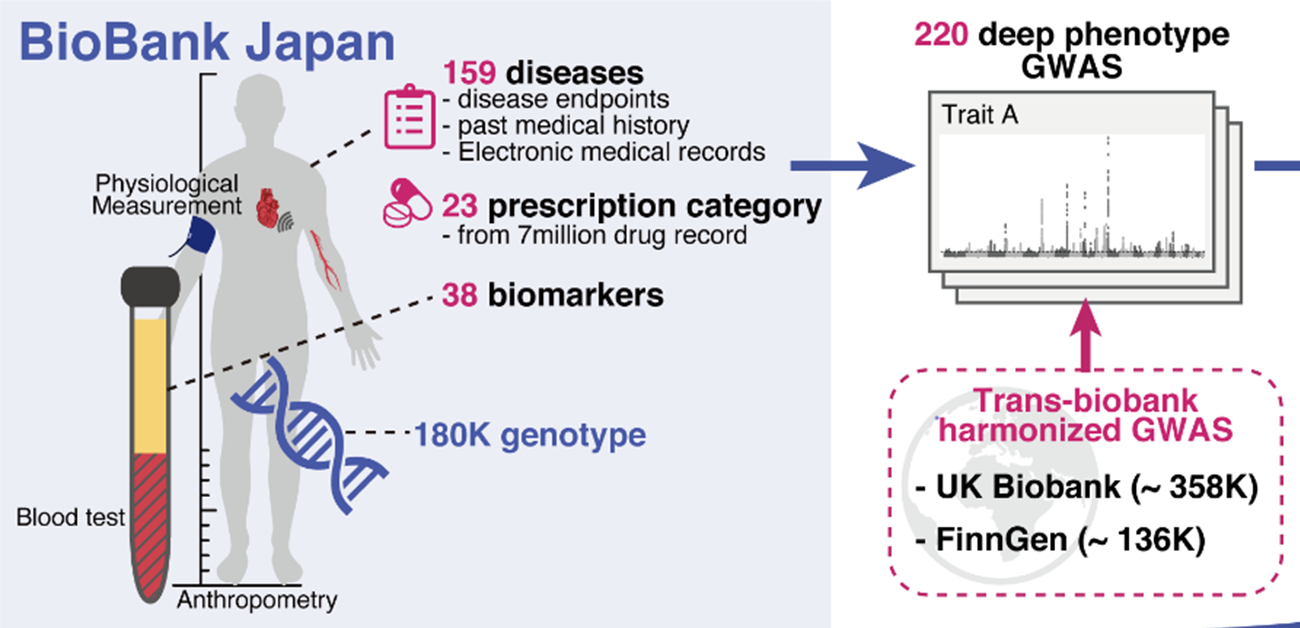
Fig.1 Overview of this study. We performed 220 deep-phenotype genome-wide association studies (GWAS) in BioBank Japan, then performed trans-biobank meta-analyses with UK Biobank and FinnGen (ntotal = 628,000). As downstream analyses, we performed (i) cross-population comparisons of pleiotropy and genetic correlations, (ii) comprehensive HLA fine-mapping, and (iii) statistical decomposition of a matrix of summary statistics to gain insights into the underlying biology of current disease classifications by incorporating functional genomics, metabolomics, and biomarker data.
(credit: © 2021 Saori Sakaue et al., Nature Genetics)
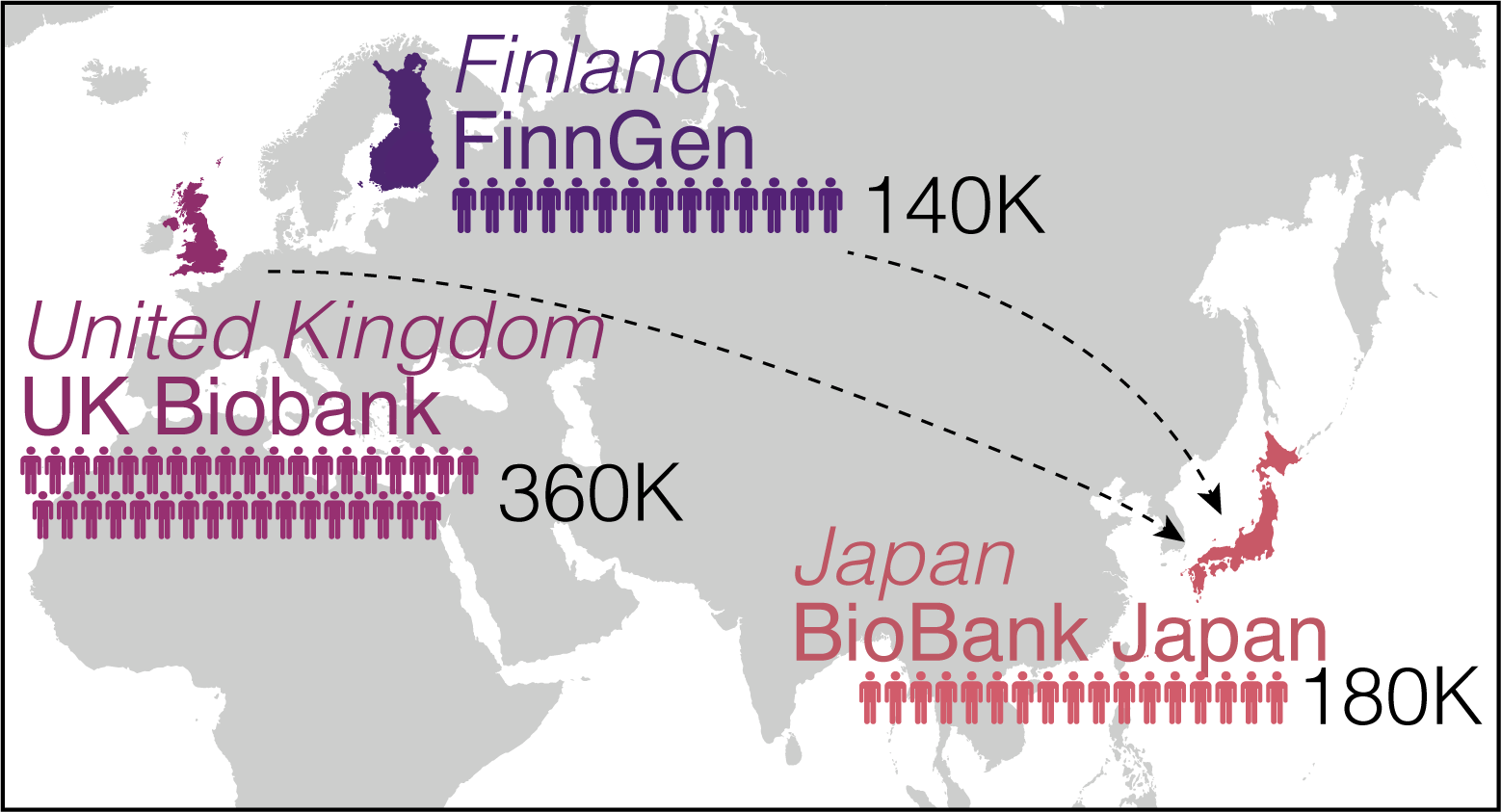
Fig.2 We studied three biobanks: BioBank Japan, UK Biobank, and FinnGen.
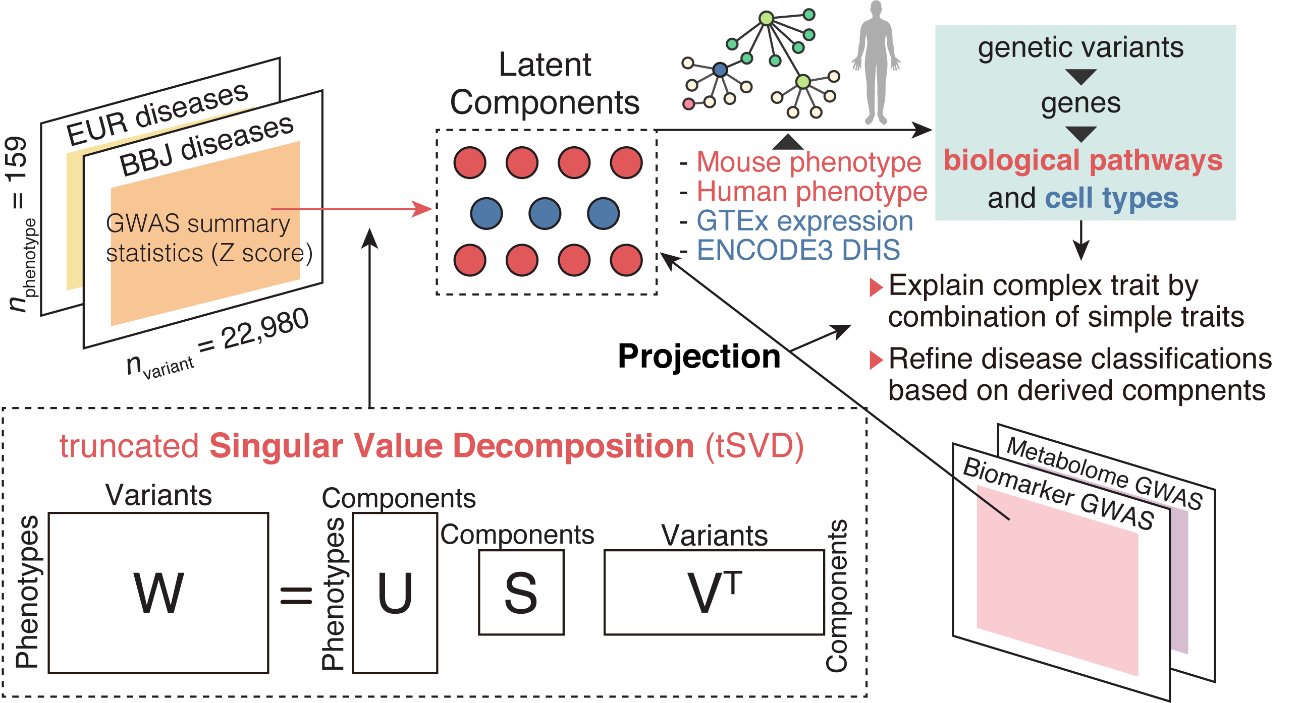
Fig.3 Using truncated singular value decomposition (TSVD), a summary statistics matrix of 159 diseases was decomposed into latent components, which were interpreted by annotating a set of genetic variants driving each component and in the context of other genome-wide association studies (GWAS) through projection.
The article, “A cross-population atlas of genetic associations for 220 human phenotypes,” was published in Nature Genetics at DOI: https://doi.org/10.1038/s41588-021-00931-x.