
Lipids, lysosomes, and autophagy: the keys to preventing kidney injury
A team of researchers led by Osaka University unravel a tangle of pathways involving calcium, lipids, and the degradation of damaged lysosomes that is essential for preventing crystal-induced kidney injury
Human cells need to work like well-oiled machines to keep our bodies running as they should. Waste products such as misfolded proteins, damaged cellular components, and carbohydrates get in the way and must be quickly disposed of. Dealing with this cellular “trash” are spherical, membrane-bound organelles called lysosomes filled with a mixture of potent enzymes. In a process called autophagy, waste products are contained within a double-membraned vesicle, called an autophagosome, that fuses with a lysosome. The lysosomal enzymes then get to work breaking down the waste into components that can be recycled.
The problem with lysosomes is that if they are ruptured, their contents can leak out and cause serious damage to the cell. Calcium oxalate crystal-induced kidney injury, which is linked to the progression of chronic kidney disease, is actually the result of lysosomal damage caused by the crystals. It is not surprising then that cells have several pathways to repair or quickly eliminate damaged lysosomes. Yet the exact steps in these pathways and how they interact during the lysosomal damage response are not entirely clear.
In a study published in Nature Cell Biology , a team of researchers led by Osaka University has finally unraveled the interactions among the lysosomal damage response pathways and determined how they prevent oxalate-induced kidney injury.
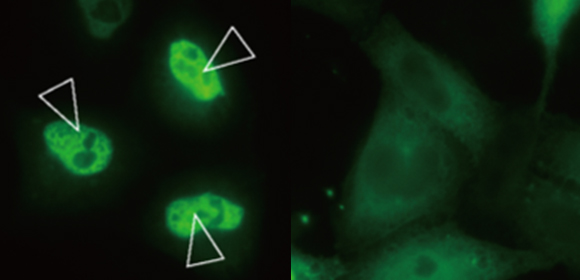
“A protein called TFEB turns on genes necessary for autophagy and the production of new lysosomes in response to lysosomal damage,” explains lead author Shuhei Nakamura. “By inhibiting TFEB function in HeLa cells and then inducing lysosome damage, we confirmed that TFEB is activated upon lysosomal damage and is necessary for the removal of damaged lysosomes.”
Attachment of lipids to a protein called LC3 is an essential step in the formation of the autophagosome. To their surprise, the researchers also found that lipidated LC3 was necessary for the activation of TFEB during the lysosomal damage response, but there was no clear link between the systems.
“Calcium is a known activator of TFEB,” says senior author Tamotsu Yoshimori. “To identify how the TFEB and LC3 systems overlapped, we investigated lysosomal calcium channel TRPML1. We found that lipidated LC3 was recruited by lysosomes in response to damage, and that the lipidated protein interacted with TRPML1, causing increased calcium efflux from the lysosome, which activated TFEB.”
The physiological importance of this interaction was then confirmed using a mouse model of oxalate crystal-induced kidney damage. Mice lacking TFEB had more severe kidney damage compared with control animals. Understanding how these pathways interact is the first step in preventing lysosomal damage-associated diseases.
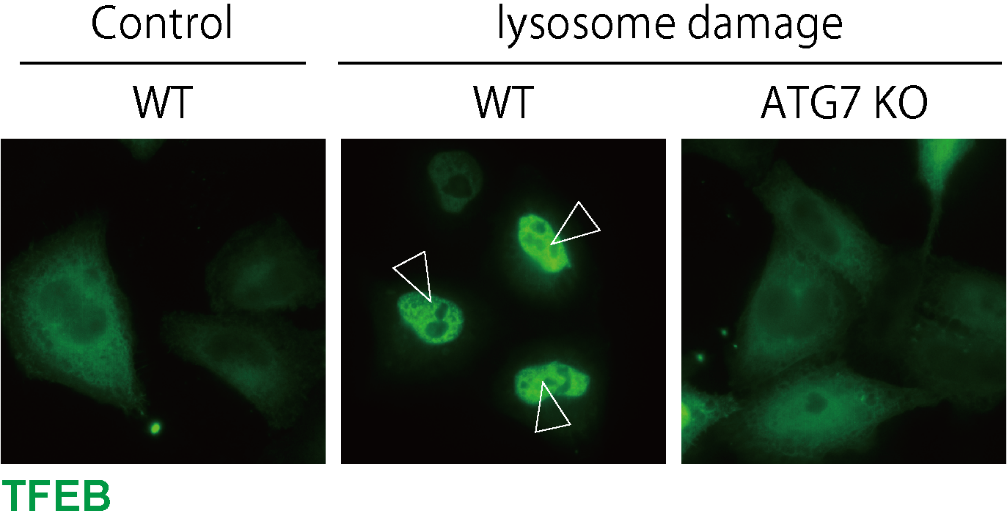
Figure 1. LC3 lipidation is essential for TFEB activation.
TFEB is localized to the nucleus upon lysosomal damage in WT cells, while ATG7 KO cells, in which LC3 lipidation is defective, show impaired TFEB nuclear localization.
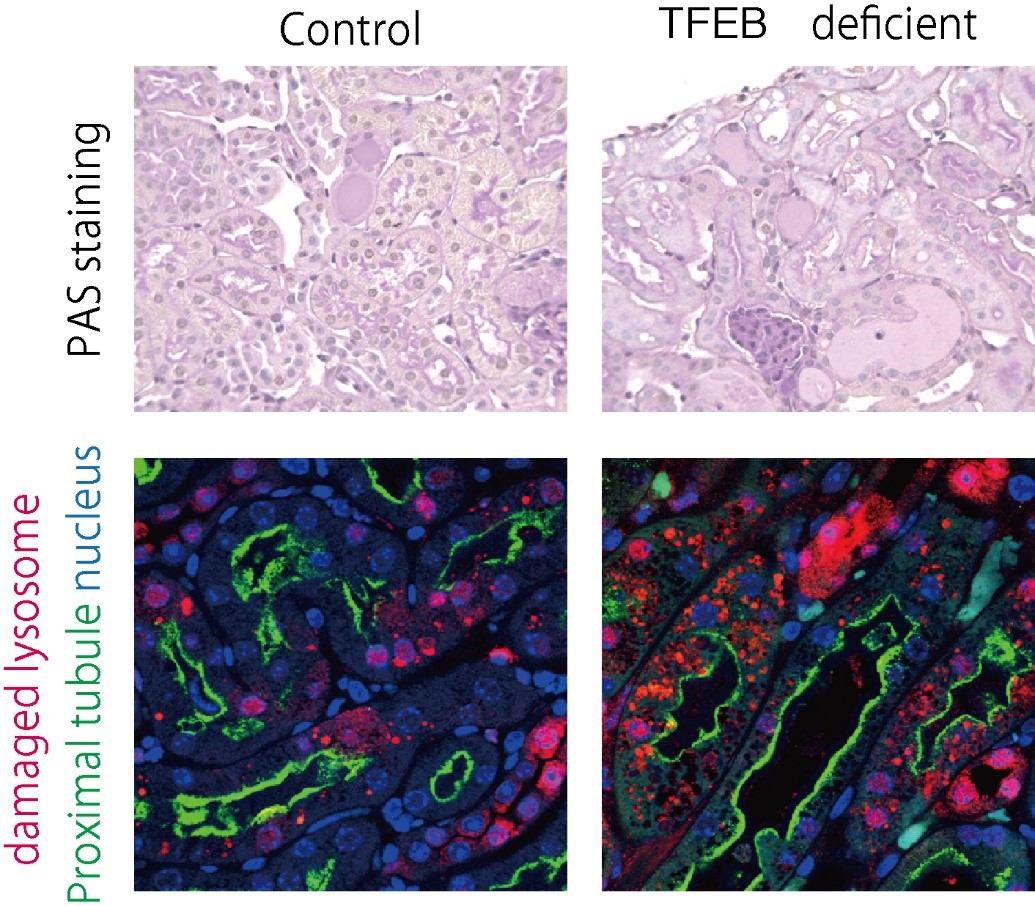
Figure 2. Functional TFEB is required to prevent the progression of crystal nephropathy.
In the crystal nephropathy mouse model, TFEB-deficient mice showed increased severity of kidney injury and lysosomal damage compared with control mice.
The article, “LC3 lipidation is essential for TFEB activation during the lysosomal damage response upon kidney injury,” was published in Nature Cell Biology at DOI: https://doi.org/10.1038/s41556-020-00583-9 .
Related Links