この研究についてひとこと
本研究では、腸内微生物叢シークエンシングデータ中に存在するヒトゲノム由来配列が、どれほどの個人情報を持っているのかを詳細に評価しました。本研究によって、腸内微生物叢研究におけるデータ共有やデータの有効活用が健全な形で促進され、医学・生物学研究の今後の発展につながることを、心より願っております。本研究は大阪大学医学部附属病院や関連施設より提供していただいたサンプルを用いることで達成することができました。全ての共同研究者や研究支援機構、並びにサンプルを提供してくださった方々に深く感謝を申し上げます。(友藤嘉彦さん)

岡田随象
- 医学系研究科
- 教授
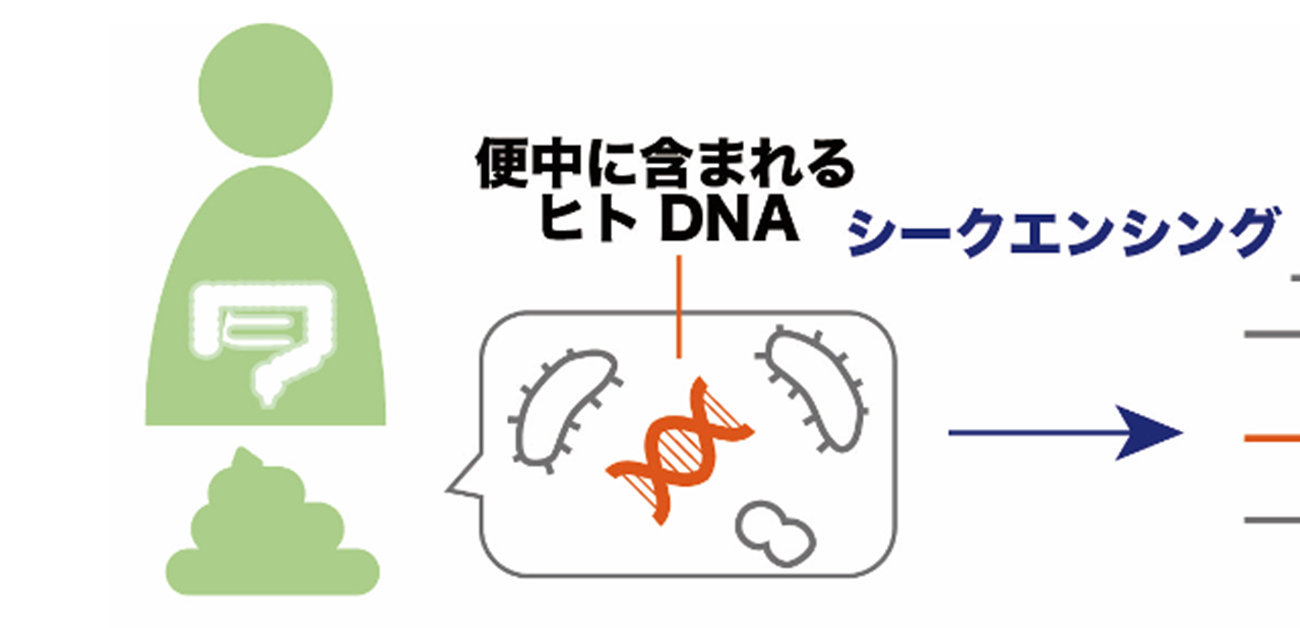
腸内微生物叢シークエンシングデータ中に存在する ヒトゲノム由来配列からの個人情報の再構築
研究成果のポイント
- 腸内微生物叢シークエンシングデータ中にわずかに存在するヒトゲノム由来配列から、性別及び属する人種集団を高精度に推定できることを示しました。
- 腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列を利用し、同一個人に由来する遺伝子多型データ・腸内微生物データの対応関係を高精度に推定できることを示しました。
- 高深度腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列から、個人の遺伝子多型情報をゲノム領域全体にわたって再構築できることを示しました。
概要
大阪大学大学院医学系研究科の大学院生の友藤嘉彦さん(遺伝統計学)、岡田随象 教授(遺伝統計学/理化学研究所生命医科学研究センター システム遺伝学チーム チームリーダー)らの研究グループは、腸内微生物叢シークエンシングデータ中に含まれるごくわずかなヒトゲノム由来配列情報に対して、新規開発手法を適用することで、性別および属する人種集団を高精度に推定できることを示しました(図1)。
また、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列を利用し、同一個人に由来する遺伝子多型データと腸内微生物叢シークエンシングデータの対応関係を高精度に推定できることを示しました。さらに、高深度に腸内微生物叢シークエンシングを行った場合、データ中に存在するヒトゲノム由来配列を用いることで、便検体から個人の遺伝子多型情報を再構築できることを示しました。
細菌やウイルスなど、数多くの微生物によって構成される腸内微生物叢は、宿主の健康状態に影響を与えることが知られています。近年の次世代シークエンシング技術の向上もあり、現在、多くの研究者達が便検体からの腸内微生物叢シークエンシング解析に取り組んでいます。腸内微生物叢シークエンシングを行うと、細菌やウイルスに由来する配列だけではなく、ごくわずかにヒトゲノム由来配列が得られることが知られていました。一般的に、遺伝子多型情報に代表されるヒトゲノム情報については、個人情報保護の観点から、慎重な取り扱いが必要とされます。しかし、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列については、その量があまりにも少なく、どれほどの個人情報が取得可能なのかが不明だったため、取り扱いについて明確な指針がないのが現状です。また、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列を有効活用できる可能性についても検討されていませんでした。
本研究成果によって、便検体及び腸内微生物叢シークエンシングデータ中に含まれるヒトゲノム由来配列を用いて、個人情報の再構築を行うことが出来ました。本研究成果は、データ共有時のプライバシーの保護や、ポリジェニック・リスク・スコアの構築などのデータの有効活用について議論する上で重要なリソースになることが期待され、健全かつ持続的な医学・生命科学研究の発展に資すると期待されます。
本研究成果は、2023年5月16日(火)午前0時(日本時間)に英国科学誌「Nature Microbiology」(オンライン)に掲載されました。
図1. 本研究の概要
研究の背景
我々の腸内には、細菌やウイルスなど、数多くの微生物が存在し、腸内微生物叢を構成しています。腸内微生物叢は免疫反応や代謝応答を介して我々の体に大きな影響を与えており、多くの医学研究の対象となっています。腸内微生物叢の解析手法には様々なものがありますが、近年の次世代シークエンシング技術の発展に伴って、便検体からの腸内微生物叢シークエンシング解析が盛んに行われるようになってきました。腸内微生物叢研究で得られたシークエンシングデータは多くの場合、公共のデータベースに登録され、世界中の研究者が誰でもアクセス可能な状態になります。研究者間でデータを共有することは研究の再現性の担保や、研究リソースの有効活用に繋がるため、医学・生命科学研究において有益と考えられますが、一方で研究参加者のプライバシーには十分に注意する必要性があります。
一般的に、遺伝子多型情報に代表されるヒトゲノム情報の公開に際しては、個人情報保護の観点から、データを慎重に取り扱うことが要求されます。腸内微生物叢シークエンシングデータ中にも約1%以下のヒトゲノム由来配列が含まれていることが知られていましたが、このごく少量のヒトゲノム由来配列からどれほどの個人情報を取得可能なのかについては不明でした。究極の個人情報とも言われる遺伝子多型情報については、通常の腸内微生物叢シークエンシングデータから再構築するのが困難であることが既に示されていましたが、高深度のシークエンシングや、同一個人由来の複数サンプルのシークエンシングを行って、通常よりも多くのヒトゲノム由来配列が得られた場合については検討されていませんでした。以上のような背景から、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列の取り扱いについては明確な規定がありませんでした。また、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列を有効活用する方法について検討されておらず、これらの配列情報は解析の対象外となっていました。
研究の成果
今回、研究グループは、腸内微生物叢シークエンシングデータから、ヒトゲノム由来配列を抽出し、どれほどの個人情報を取得できるのかについて評価を行いました。
まず、研究グループは、腸内微生物叢シークエンシングデータに含まれるヒトゲノム由来配列のうち、ヒトのX・Y染色体に由来するものを利用して、性別の推定を行いました。343名の訓練用データセットを用いて訓練されたロジスティック回帰モデルを、113名の検証用データセットに適用したところ、97.3%の正答率で性別を予測することに成功しました。
次に、研究グループは同一個人から取得した腸内微生物叢シークエンシングデータと遺伝子多型データとを用いて、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列と、同一個人に由来する遺伝子多型データとを紐づけられるかどうか、検討しました(図2)。研究チームは腸内微生物叢シークエンシングデータと遺伝子多型データのペアについて、2つのデータが同一個人由来の時に高い値をとる、尤度スコアを導入しました。その後、343名の腸内微生物叢シークエンシングデータと遺伝子多型データを用いて、実際に尤度スコアに基づいた同一個人予測を行ったところ、93.3%の正答率が得られました。
図2. 尤度スコアに基づいた、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列と、同一個人に由来する遺伝子多型データとの紐付け
さらに、研究グループは、個人がどの人種集団に属するのかを予測するために、腸内微生物叢シークエンシングデータが特定の人種集団(例:東アジア人集団、ヨーロッパ人集団等)に由来する時に高い値を取る尤度スコアを導入しました(図3)。実際に、様々な人種集団に由来する腸内微生物叢シークエンシングデータに対して、尤度スコアに基づいた予測を適用したところ、人種集団によってばらつきがあるものの、80〜98%の正答率が得られました。
図3. 尤度スコアに基づいた、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列からの人種集団予測
最後に、研究グループは、高深度腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列から、遺伝子多型情報を取得しました。高深度腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列の量は、一般的なヒト全ゲノムシークエンシングなどと比較して少ないため、研究チームはtwo-step imputation法によって、外部の参照ゲノム配列データを利用し、集団中に比較的高頻度に存在する遺伝子多型(コモンバリアント)情報をゲノム領域全体にわたって再構築しました。また、外部の参照データを利用せずに遺伝子多型情報を取得した場合には、ゲノム領域全体の情報を得るのは難しいものの、一部の集団中にごく低頻度にしか存在しない遺伝子多型(レアバリアント)の情報を取得できることもわかり、それらの一部は過去に希少難病疾患との関連が報告されている遺伝子上に位置する多型でした。
本研究成果が社会に与える影響(本研究成果の意義)
本研究成果によって、腸内微生物叢シークエンシングデータ中に含まれるヒトゲノム由来配列から、様々な個人情報を抽出できることがわかりました。今回開発した手法を用いることで、以前は解析対象外となっていた、腸内微生物叢シークエンシングデータ中のヒトゲノム由来配列情報を有効活用することが可能になり、特に、法医学分野での活用や、ポリジェニック・リスク・スコア構築をはじめとした、個別化医療への応用が期待されます。本研究成果は、データ共有に際するプライバシーの保護や、データの有効活用について議論する上で重要なリソースになることが期待され、健全かつ持続的な医学・生命科学研究の発展に資すると期待されます。
特記事項
本研究成果は、2023年5月16日(火)午前0時(日本時間)に英国科学誌「Nature Microbiology」(オンライン)に掲載されました。
【タイトル】 “Reconstruction of the personal information from human genome reads in gut metagenome sequencing data”
【著者名】Yoshihiko Tomofuji1,2,3*, Kyuto Sonehara1,2,4, Toshihiro Kishikawa1,5,6, Yuichi Maeda2,7,8, Kotaro Ogawa9, Shuhei Kawabata10, Takuro Nii7,8, Tatsusada Okuno9, Eri Oguro-Igashira7,8, Makoto Kinoshita9, Masatoshi Takagaki10, Kenichi Yamamoto1,11,12, Takashi Kurakawa8, Mayu Yagita-Sakamaki7,8, Akiko Hosokawa9,13, Daisuke Motooka2,14, Yuki Matsumoto14, Hidetoshi Matsuoka15, Maiko Yoshimura15, Shiro Ohshima15, Shota Nakamura2,14,16, Hidenori Inohara5, Haruhiko Kishima10, Hideki Mochizuki9, Kiyoshi Takeda8,16,17, Atsushi Kumanogoh2,7,18,19, Yukinori Okada1,2,3,4,12,16,19*(*責任著者)
【所属】
1. 大阪大学大学院医学系研究科 遺伝統計学
2. 大阪大学先導的学際研究機構(OTRI) 生命医科学融合フロンティア研究部門
3. 理化学研究所 生命医科学研究センター システム遺伝学チーム
4. 東京大学大学院医学系研究科 遺伝情報学
5. 大阪大学大学院医学系研究科 耳鼻咽喉科・頭頸部外科学
6. 愛知県がんセンター 頭頸部外科部
7. 大阪大学大学院医学系研究科 呼吸器・免疫内科学
8. 大阪大学大学院医学系研究科 免疫制御学
9. 大阪大学大学院医学系研究科 神経内科学
10. 大阪大学大学院医学系研究科 脳神経外科学
11. 大阪大学大学院医学系研究科 小児科学
12. 大阪大学 免疫学フロンティア研究センター(IFReC) 免疫統計学
13. 吹田市民病院 脳神経内科
14. 大阪大学 微生物病研究所 感染症メタゲノム研究分野
15. 大阪南医療センター リウマチ・膠原病・ アレルギー科
16. 大阪大学 感染症総合教育研究拠点(CiDER)
17. 大阪大学 免疫学フロンティア研究センター(IFReC) 粘膜免疫学
18. 大阪大学 免疫学フロンティア研究センター(IFReC) 感染病態分野
19. 大阪大学 先端モダリティ・ドラッグデリバリーシステム研究センター(CAMaD)
DOI:https://doi.org/10.1038/s41564-023-01381-3
本研究は、日本医療研究開発機構(AMED)ゲノム医療実現推進プラットフォーム事業・先端ゲノム研究開発(GRIFIN)の採択課題「遺伝統計学に基づく日本人集団のゲノム個別化医療の実装」(研究開発代表者:岡田随象)の一環として行われ、大阪大学免疫学フロンティア研究センター 次世代主任研究者支援プログラム、大阪大学先導的学際研究機構、大阪大学大学院医学系研究科 バイオインフォマティクスイニシアティブ、武田科学振興財団の協力を得て行われました。
用語説明
- 人種集団
本研究においては、共通の遺伝学的特徴を持つ人々の集まりのことを指し、国際1,000人ゲノムプロジェクト(URL:http://www.1000genomes.org)で用いられた定義を用いている。
- 腸内微生物叢
宿主であるヒトや動物と共生関係にある多種多様な腸内微生物の集まり。
- 次世代シークエンシング
数千から数百万ものDNA分子を同時に配列決定する手法。
- 腸内微生物叢シークエンシング
微生物の全ゲノムDNAを短いDNA鎖に切断してライブラリを作成し、次世代シークエンサーによって配列決定する手法。
- 遺伝子多型
遺伝子を構成している塩基配列の個体差。一塩基多型(Single Nucleotide Polymorphism; SNP)などが代表的。
- ポリジェニック・リスク・スコア
ヒトゲノム配列上に存在する数百万カ所の遺伝子多型のうち、疾患との関連が示唆された数十〜数十万の遺伝子多型について、効果量の重み付きの和を個人ごとに計算したスコア。このスコアは疾患発症リスクと相関することが知られている。
- ロジスティック回帰モデル
目的変数が2値のデータ(今回は性別)を、説明変数(今回はX・Y染色体由来の配列の比率)を使った式で表す方法。
- 尤度スコア
本研究中で定義された、メタゲノムショットガンシークエンスデータ中のヒトゲノム由来配列と個人のSNP情報をもとに計算されるスコア。尤度スコアは、SNP情報の由来する個人と、腸内微生物叢シークエンシングデータの由来する個人とが同一であるという事象の起こりやすさを反映している。集団中のアレル頻度情報を用いることで、SNP情報の由来する個人と、腸内微生物叢シークエンシングデータの由来する個人とが異なる場合の尤度スコアの分布を推定でき、この分布と比較を行うことで、実際に得られた尤度スコアがどれほど高いのかをP値として評価することができる。
- two-step imputation法
まず1段階目として、シークエンシングデータ中のヒトゲノム由来配列がカバーしているゲノム領域について、参照ゲノム配列データを利用して、遺伝子型を決定する。その後、1段階目では決定できなかった遺伝子多型の情報を、ゲノム配列上で周囲に位置する遺伝子多型の情報と参照ゲノム配列データに基づいて推定する。