
New drug targets for a rare kidney and liver disease
In a joint international study, researchers from Osaka University have partnered with research groups from the United States and Spain to uncover how mutations in a single gene called PKHD1 lead to symptoms associated with a rare kidney and liver disease, ARPKD (autosomal recessive polycystic kidney disease). The findings are expected to lead to novel treatment strategies against the disease.
ARPKD is a rare disease affecting 1 in 20000 people, but its victims are newborns, many of whom fail to see their first birthday. As for those who do survive, they often suffer from a litany of ailments including hypertension, cyst formation, and tissue scarring. Treatments normally target the symptoms of the disease because the underlying molecular causes are poorly understood.
ARPKD is caused by a mutation in the PKHD1 gene. To identify key candidate molecules that contribute to the disease, Osaka University Associate Professor Jun-Ya Kaimori and his colleagues compared cells from Pkhd1 mutant and normal mice, finding that the different expression levels of the Pkhd1 gene correlated with cell shape and cyst formation.
“ PKDH1 encodes for fibrocystin/polyductin complex (FPC). The altered cell morphologies depended on the level of RhoA,” explains Kaimori.
RhoA is protein that regulates cell morphology. The researchers therefore looked for molecular mechanisms through which RhoA and FPC interact, setting their eyes on ubiquitin ligases that regulate RhoA. They found that in normal mice, three E3 family members of ubiquitin ligases colocalized with FPC, but in mutant mice, the three were abnormally distributed in the cells. Adding to the importance of this observation is that the three ligases are known to have roles in regulating the three major symptoms of ARPKD.
“We found that reducing the activity of FPC altered the localization of three different E3 family ligases that contribute to cyst genesis, liver and kidney fibrosis, and hypertension,” says Kaimori.
However, Kaimori doubts only these three ligases are affected by the mutation.
“Our results suggest that the disruption of Pkhd1 and FPC is likely to alter the activity of other E3 ligases,” he says.
Clarifying these other ligases will provide more detail on the wide range of ailments that inflict ARKPD patients and provide promising targets for future medicines.
Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is an important childhood nephropathy, occurring 1 in 20,000 live births. The major clinical phenotypes are expressed in the kidney with dilatation of the collecting ducts, systemic hypertension, and progressive renal insufficiency, and in the liver with biliary dysgenesis, portal tract fibrosis, and portal hypertension. The systemic hypertension has been attributed to enhanced distal sodium reabsorption in the kidney, the structural defects have been ascribed to altered cellular morphology, and fibrosis to increased TGF-β signaling in the kidney and biliary tract, respectively. The pathogenic mechanisms underlying these abnormalities have not been determined. In the current report, we find that disrupting PKHD1 results in altered sub-cellular localization and function of the C2-WWW-HECT domain E3 family of ligases regulating these processes. We also demonstrate altered activity of RhoA and increased TGF-β signaling and ENaC activity. Linking these phenomena, we found that vesicles containing the PKHD1/Pkhd1 gene product, FPC, also contain the NEDD4 ubiquitin ligase interacting protein, NDFIP2, which interacts with multiple members of the C2-WWW-HECT domain E3 family of ligases. Our results provide a mechanistic explanation for both the cellular effects and in vivo phenotypic abnormalities in mice and humans that result from Pkhd1/PKHD1 mutation.
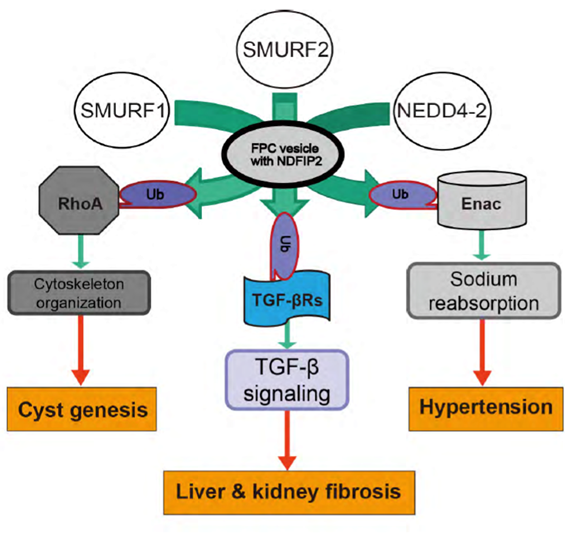
Figure 1
To learn more about this research, please view the full research report entitled " NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology " at this page of the Scientific Reports website.
Related links