
Cause of heart arrhythmia in adult myodystrophia clarified
Will lead to the prevention of death in this disease and the development of treatment methods
An international joint research team including Professor TAKAHASHI Masanori (Graduate School of Medicine, Osaka University) discovered that the cause of heart arrhythmia in myotonic dystrophy (DM), the most common muscular dystrophy in adults, was RNA abnormalities in the sodium channel in the heart, clarifying the symptom’s mechanism.
There are no treatment methods for DM and there are many cases of death suspected to be due to heart arrhythmia. So, the clarification of the cause of heart arrhythmia will be helpful in prevention and early intervention of death in this disease, leading to the development of new treatment methods.
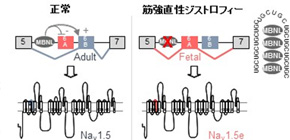
RNA abnormalities have drawn attention as the cause of manifestation of various systemic conditions of DM. That is, abnormally extended RNA tends to abnormally accumulate in the nucleus and takes proteins necessary for RNA splicing that takes place within the nucleus. Therefore, it is thought that various RNA splicing cannot be controlled, which causes abnormalities of proteins, manifesting various conditions. For example, in 2002, Prof. Takahashi suggested that muscle stiffness, a feature of DM, represented decreased proteins in chloride ion channels due to splicing abnormalities, but the cause of heart arrhythmia has been unknown for a long time.
This group found that there were RNA splicing abnormalities in the sodium channel in DM patients. This group electrophysiologically examined the function of an abnormal sodium channel and found that it was compromised. From this finding, it was suggested that patients manifested conditions similar to Brugada syndrome, which led to their fatal heart arrhythmia.
When this group caused RNA abnormalities manifested in patients in mice, heart arrhythmia was observed. In addition, a computer simulation demonstrated that the compromised sodium channel function caused abnormalities in an electrocardiogram, which was similar to abnormalities in patients.
From these findings, it was concluded that heart arrhythmia and death in DM patients were caused by RNA splicing abnormalities in the sodium channel. The clarification of the cause of heart arrhythmia in DM will lead to the prediction of the progress of and early intervention for heart arrhythmia, preventing death in this disease.
Repeat diseases include DM and other neurodegenerative disorders. Therefore, this group’s research method and the idea of pathological mechanisms involving RNA abnormalities will be applied to pathological research on various diseases.
Abstract
Myotonic dystrophy (DM) is the most common muscular dystrophy in adults, and the first recognized example of a RNA-mediated disease. DM is caused by the expression of mutant RNAs containing expanded CUG or CCUG repeats. These mutant RNAs sequester muscleblind-like (MBNL) splicing regulators, leading to specific alternative splicing changes that ultimately result in the symptoms of myotonic dystrophy. Cardiac alterations, characterized by conduction delays and arrhythmia, are the second most common cause of death in DM. Using massive parallel RNA sequencing, we identified novel splicing alterations in heart samples of DM patients, among which, a switch from inclusion of the adult exon 6B toward usage of the fetal exon 6A of the cardiac sodium channel, SCN5A. Mutations in SCN5A cause a variety of arrhythmic disorders, which present similar features to cardiac alterations observed in myotonic dystrophy. We found that MBNL1 regulates alternative splicing of SCN5A and that depletion of MBNL activities reproduces the misregulation of SCN5A splicing observed in cardiac samples of DM patients. Next, we confirmed that the splicing variant of SCN5A produced in DM, namely hNav1.5e, presents a reduced excitability compared to hNav1.5, which is encoded by the normal adult isoform of SCN5A. Importantly, reproducing this specific splicing alteration of Scn5a in wild-type mice is sufficient to promote heart arrhythmia and cardiac conduction delay, two predominant features of myotonic dystrophy. A result confirmed by mathematical simulation. In conclusion, misregulation of the alternative splicing of SCN5A in DM results in expression of a fetal splicing form of the cardiac sodium channel, which has altered electrophysiological properties and leads to cardiac arrhythmia and conduction delay in mice. We propose that these findings may contribute to the cardiac dysfunctions observed in DM patients.

Model of SCN5A splicing alteration in myotonic dystrophy.
MBNL proteins regulate the switch from exon 6A in embryonic heart to exon 6B in adult. In myotonic dystrophic patients, titration of MBNL protein by RNA containing expanded CUG repeats leads to expression of a splicing form of the cardiac sodium channel, SCN5A, inappropriate to adult heart physiology, ultimately resulting in cardiac conduction delay and heart arrhythmias, which are two keys features of myotonic dystrophy.
To learn more about this research, please view the full research report entitled " Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy " at this page of the Nature Communications website.
Related link