
Pathogenic mechanism of cardiac failure after acute myocardial infarction clarified
Towards the prevention of cardiac failure in the chronic phase
The onset of cardiac failure after acute myocardial infarction (AMI) is a serious problem throughout the world.
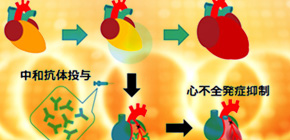
Researchers at Osaka University clarified that the cell adhesion inhibition of periostin1 damages myocardinal cells, inducing compromised cardiac myocyte contractile force and myocytes death, leading to the onset of cardiac failure after AMI through the administration of periostin neutralizing antibodies they had developed on their own.
If the onset of cardiac failure in the chronic phase can be inhibited by this group’s research results that will not only improve patients’ quality of life, but also reduce high medical costs of cardiac failure treatment as a whole.
Yoshiaki Taniyama, Endowed Chair Associate Professor, Ryuichi Morishita, Endowed Chair Professor, and Fumihiro Sanada, Specially Appointed Assistant Professor at Osaka University focused their attention on periostin, a protein secreted mainly from fibroblasts by mechanical stretch (mechanical stress) after the onset of AMI.
Acute myocardial infarction generates mechanical stress on the heart, which causes secretion of periostin from fibroblastsand promotes cardiac failure (Figure 1); however, its mechanism was unclear. There are four splice variants of periostin gene, and Associate Professor Taniyama et al. have reported that periostin1 solely induced cardiac failure. Furthermore, researchers at Harvard University, the University of Cincinnati, and Tokyo Institute of Technology reported that inhibition of all periostins suppressed cardiac failure after AMI, but periostin2 and periostin4 had cardioprotective effects such as myocardial regeneration, angiogenesis, and protection of cardiac rupture after AMI. The suppression of all periostin variants inhibits cardiac failure, but increases death by cardiac rupture in the acute phase after AMI.
Thus, this group considered a therapeutic method in which not periostin2 and periostin4 with cardioprotective effects but periostin1 with cardiac failure -inducing effects was suppressed. This group independently developed periostin1-specific neutralizing antibodies and administered them to patients after the onset of acute myocardial infarction, thereby succeeding in inhibiting the onset of cardiac failure in the chronic phase without the accompaniment of cardiac rupture.
Abstract
We previously reported that overexpression of full-length periostin, Pn-1, resulted in ventricular dilation with enhanced interstitial collagen deposition in a rat model. However, other reports have documented that the short-form splice variants Pn-2 (lacking exon 17) and Pn-4 (lacking exons 17 and 21) promoted cardiac repair by angiogenesis and prevented cardiac rupture after acute myocardial infarction. The apparently differing findings from those reports prompted us to use a neutralizing antibody to selectively inhibit Pn-1 by blockade of exon 17 in a rat acute myocardial infarction model. Administration of Pn neutralizing antibody resulted in a significant decrease in the infarcted and fibrotic areas of the myocardium, which prevented ventricular wall thinning and dilatation. The inhibition of fibrosis by Pn neutralizing antibody was associated with a significant decrease in gene expression of fibrotic markers, including collagen I, collagen III, and transforming growth factor-β1. Importantly, the number of α-smooth muscle actin–positive myofibroblasts was significantly reduced in the hearts of animals treated with Pn neutralizing antibody, whereas cardiomyocyte proliferation and angiogenesis were comparable in the IgG and neutralizing antibody groups. Moreover, the level of Pn-1 expression was significantly correlated with the severity of myocardial infarction. In addition, Pn-1, but not Pn-2 or Pn-4, inhibited fibroblast and myocyte attachment, which might account for the cell slippage observed during cardiac remodeling.
Collectively, these results indicate that therapeutics that specifically inhibit Pn exon-17, via a neutralizing antibody or drug, without suppressing other perisotin variants might offer a new class of medication for the treatment of acute myocardial infarction patients.

Fig1: Congestive heart failure (CHF) after acute myocardial infarction
When acute myocardial infarction (AMI) occurs, mechanical stress increases periostin which promotes congestive heart failure (CHF), but the mechanism was not clear.
By the way, there are 4 splice variants in periostin, and Dr. Taniyama reported that only Pn-1 overexpression in heart promotes CHF. The other people reported that Pn-2 promotes angiogenesis and myogenesis, Pn-4 protects cardiac rupture after AMI, and ablation of Pn1-4 in heart protects CHF appearance after AMI with the increase in cardiac rupture at acute phase. In addition, Dr. Taniyama showed that Pn-1 but not Pn-2 or Pn-4 increases myocyte death through inhibiting cell attachment. Getting together, we think that Pn-1 but not Pn-2 or Pn-4 should be inhibited after AMI. We made the Pn-1 specific neutralizing antibody, and its treatment protects CHF appearance after AMI without cardiac rupture.
Periostin -- A secreted extracellular matrix protein that functions in tissue development , tissue remodeling. Alternative splicing results in multiple transcript variants encoding almost 4 isoforms.
Mechanical stress -- physical load which activates cell signaling
Alternative Splicing -- A regulated process during gene expression that results in a single gene coding for multiple proteins with the lack of particular exon.
To learn more about this research, please view the full research report entitled " Selective Blockade of Periostin Exon 17 Preserves Cardiac Performance in Acute Myocardial Infarction " at this page of the Hypertension website.