
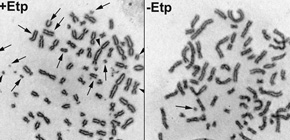
DNA repair mechanisms do not always result in effective repair and cell protection
Specific measures may be developed to prevent such genome instability
A group of researchers led by TERASAWA Masahiro (Specially Appointed Researcher) and SHINOHARA Miki (Associate Professor, Institute for Protein Research, Osaka University) discovered in experiments using human cells, that DNA repair mechanisms during mitosis at times induced genome instability.
When genetic information is broken, rewritten, or mistakenly transcribed, cells may start multiplying and become cancerous. DNA repair mechanism functions to prevent normal cells from becoming cancerous by repairing DNA damage from radiation and other causes. Because genome instability is thought to cause normal cells to become cancerous, activation of DNA repair mechanisms during mitosis is thought to cause cancer due to its resulting inappropriate and/or inadequate repair.
Thus, in normal cells, there are systems to inactivate DNA repair mechanism during mitosis. As one such system, this group has clarified that M-phase-specific phosphorylation of XRCC4 suppressed the M-phase DNA repair mechanism. Not surprisingly, the group also found that when phosphorylation was prevented, DNA double-strand breaks (DSBs) during mitosis could be repaired, but severe genome instability occurred.
Severe genome instability leads to cell death while less severe instability may induce cancerization. Cancer drugs make use of such effects. However, it is also known that chemotherapy using cancer drugs may induce secondary cancers. This group is of the opinion that DNA damage caused by cancer drugs triggers DNA repair mechanisms during mitosis, causing genome instability and thereby leading to secondary cancers.
Thus, if the DNA repair mechanism of cancer cells can be activated during mitosis, this may be utilized to increase the effectiveness of chemotherapy drugs. Furthermore, if secondary cancers caused by anticancer drug treatment are due to genome instability during mitosis, it may become possible to take specific measures to prevent such genome instability.
Abstract
DNA double-strand breaks (DSBs) can be repaired by one of two major pathways—non-homologous end-joining (NHEJ) and homologous recombination (HR)—depending on whether cells are in G1 or S/G2 phase, respectively. However, the mechanisms of DSB repair during M phase remain largely unclear. In this study, we demonstrate that transient treatment of M-phase cells with the chemotherapeutic topoisomerase inhibitor etoposide induced DSBs that were often associated with anaphase bridge formation and genome instability such as dicentric chromosomes. Although most of the DSBs were carried over into the next G1 phase, some were repaired during M phase. Both NHEJ and HR, in particular NHEJ, promoted anaphase-bridge formation, suggesting that these repair pathways can induce genome instability during M phase. On the other hand, C-terminal-binding protein interacting protein (CtIP) suppressed anaphase bridge formation, implying that CtIP function prevents genome instability during mitosis. We also observed M-phase-specific phosphorylation of XRCC4, a regulatory subunit of the ligase IV complex specialized for NHEJ. This phosphorylation required cyclin-dependent kinase (CDK) activity as well as polo-like kinase 1 (Plk1). A phosphorylation-defective XRCC4 mutant showed more efficient M-phase DSB repair accompanied with an increase in anaphase bridge formation. These results suggest that phosphorylation of XRCC4 suppresses DSB repair by modulating ligase IV function to prevent genome instability during M phase. Taken together, our results indicate that XRCC4 is required not only for the promotion of NHEJ during interphase but also for its M-phase-specific suppression of DSB repair.
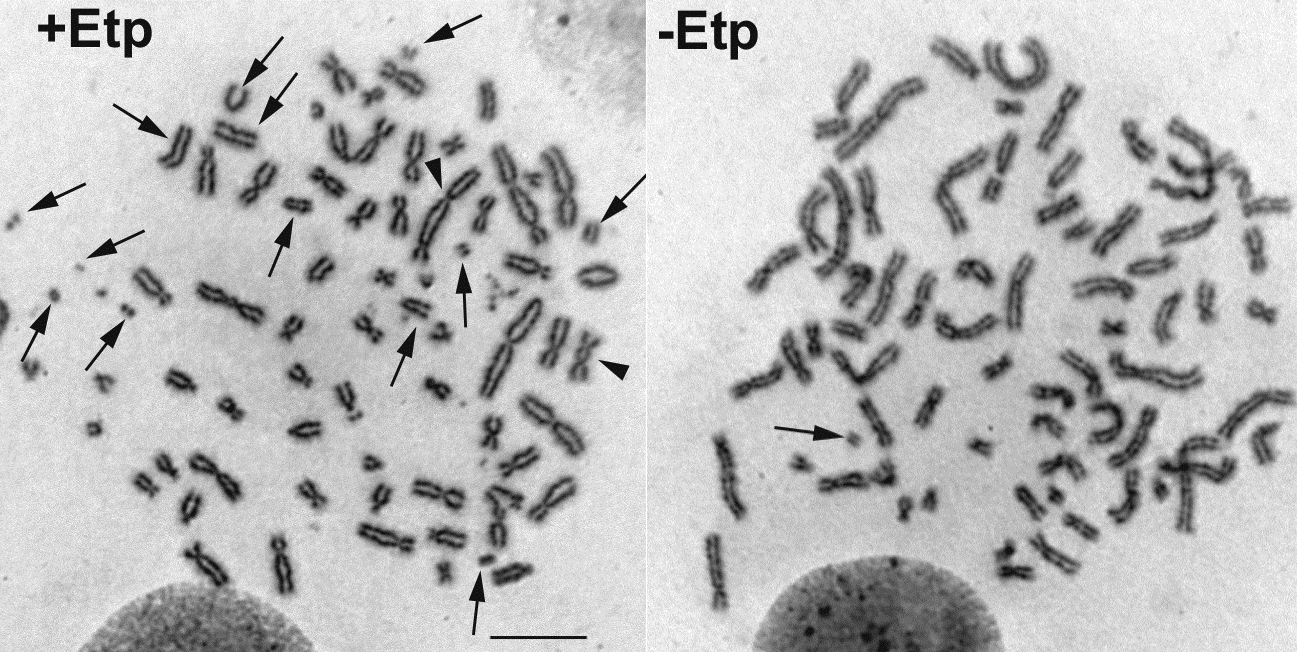
To learn more about this research, please view the full research report entitled " Canonical Non-homologous End Joining in Mitosis Induces Genome Instability and Is Suppressed by M-phase-specific Phosphorylation of XRCC4 " at this page of the PLOS Genetics website.
Related links :