
磁性分子中の複雑な電子スピン状態の新たな高精度計算手法を開発
分子スピントロニクス素子開発への応用に向けて
成果のポイント
・これまで、磁性分子中の電子スピン状態の理論的解析は、軌道の配位子場分裂 や電子間にはたらく多体的な相関効果 の複雑性により困難だった。
・本研究で開発した第一原理計算 により高精度な解析が可能に。
・磁性分子材料を用いたスピントロニクス素子の理論的設計・開発への応用に期待。
概要
大阪大学産業科学研究所の小口多美夫教授、三重大学大学院工学研究科の名和憲嗣大学院生と同大学の中村浩次准教授、ウィスコンシン大学のWeinert(ワイナート)教授らの研究グループは、磁性分子材料の電子及びスピン状態を高精度に解析・予測するための第一原理計算手法の開発に成功しました。磁性分子が持つ物性の起因となるd軌道やf軌道の電子スピン状態は、従来の第一原理的手法では計算精度に信頼性を欠く場合があり、高精度な磁性分子の物性予測を可能とする第一原理計算手法の開発が求められていました。本成果により、分子スピントロニクス素子開発の発展に向けた新規磁性分子素子を予測するための、重要な理論的手法が確立されました。
研究の背景・内容
電子が持つ二つの自由度である電荷とスピンの両者を組み合わせてデバイスへと応用するスピントロニクス技術の研究は、近年、精力的に行われており、高速且つ低消費電力を兼ね備えた素子が実現できるとして注目されています。中でも金属元素を含む磁性分子により構成される素子は、単一分子レベルで固有の機能を担うことができることから更なる小型化も期待され、分子スピントロニクスは、究極の磁性材料としての関心が高まっています。この技術を応用した高性能素子の設計・開発には、磁性分子が持つ性質の起源となる電子スピン状態を理解することが不可欠であり、第一原理計算を用いた理論的アプローチが求められます。一方、その電子スピン状態に関する第一原理計算において、電子軌道の配位子場分裂や電子間にはたらく相関効果の複雑性のために、密度汎関数理論(DFT) の適用に限界があることが知られています。実際、基底状態の電子スピン状態でさえも、実験と理論計算との両者間で整合性がとれない場合が多く、高い精度での磁性分子材料の理論的設計を行うためには、これらの障害を克服することが求められていました。
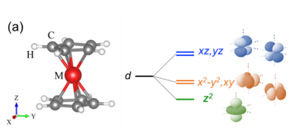
共同研究グループは、独自に開発してきたFLAPW法 を用い、考えられるあらゆる電子スピン状態( 図1 (b))の全エネルギー計算を行うことで、高精度に電子スピン状態を解析し、基底状態を探索することができる新たな第一原理計算手法の開発を行いました。さらに、電子間相関効果の取り扱いを工夫し、相関効果の程度を示す有効オンサイトクーロン相互作用力を定量的に導出し、DFT+U法 に基づき多体効果を考慮しました。一般的に、この相互作用力は定性的または経験的に与えられることが多く、それ故、誤った性質の予測に繋がるというリスクがありました。さらに、同手法をメタロセン分子( 図1 (a)) に適用した結果、安定な電子スピン状態やその起源を、世界で初めて、理論的に解明することに成功し、磁性分子材料における種々の物性予測のための新たな計算手法を確立しました。
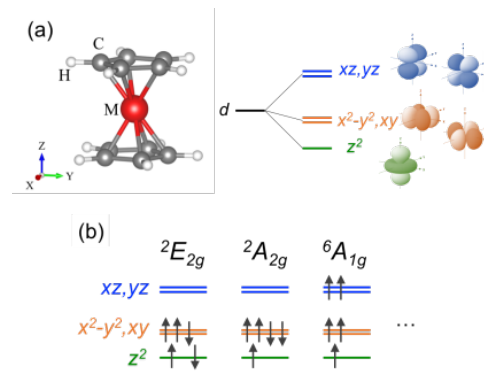
図1 (a)メタロセン分子構造と配位子場分裂した遷移金属のd軌道。(b)Mn原子を含む系(d 5 )における考えられる電子スピン状態の一例。
本研究成果が社会に与える影響(本研究成果の意義)
今回の研究は、単一磁性分子の電子スピン状態を高精度に解析し、それに起因する種々の性質や現象を予測することを可能とした理論計算手法を開発したものです。今後、単分子磁石を用いた不揮発性分子メモリや量子コンピュータ等の実用的な磁性分子素子の研究開発への展開が期待されます。
特記事項
本研究は物質・デバイス領域共同研究拠点:人・環境と物質をつなぐイノベーション創出ダイナミック・アライアンスにおける次世代若手共同研究(受入教員:大阪大学産業科学研究所小口多美夫教授)の一環として行われました。
研究者のコメント
コンピュータを用いた理論的な材料予測・設計は、次世代を担う新たなデバイス創出への指針を与える、極めて重要な役割を担っています。従って、計算精度の高さは、予測された材料の実用化の可能性と直結する指標であり、分子スピントロニクス素子開発に向けて、本研究で開発した手法の今後の応用が期待されます。
参考URL
大阪大学 産業科学研究所 産業科学ナノテクノロジーセンター ナノ機能予測研究分野
http://www.cmp.sanken.osaka-u.ac.jp/index_jp.html
用語説明
- 配位子場分裂
磁性分子中において、金属元素が持つd(f)軌道及びその周りに配位する化合物(配位子)の軌道との相互作用により軌道が分裂すること。この分裂した軌道に金属元素のd(f)電子は占有する。電子占有の仕方は、軌道の分裂幅やHund則、電子間相関効果の強さによって決まる。
- 相関効果
多電子系において電子間にはたらく相互作用のこと。厳密には、電子相関エネルギーは実際のエネルギー及びハートリー・フォックエネルギーとの差として定義される。金属元素付近に電子が局在する系ではこの差が大きくなり、一電子近似を基として構築される密度汎関数理論では、十分な計算精度が保証されない問題が生じる。
- 第一原理計算
量子力学の基本法則に基づき得られる電子状態から物質の諸性質を数値的に計算する手法。実験データや経験的パラメータは使用しない。
- 密度汎関数理論(DFT)
電子系のエネルギーを電子密度の汎関数として表す理論。実際には多電子中を運動する電子を、有効的なポテンシャルを感じて運動する一つの電子と仮定している(一電子近似)。DFTは、この近似に基づく交換相関項を導入することで、多体問題を一体問題に帰着した理論である。
- FLAPW法
全電子フルポテンシャル線形化補強平面波法。価電子及び内殻の電子状態も取り扱うことができ、且つ、原子核に由来するポテンシャルに対して球対称な近似(マフィンティン近似と呼ばれる)を排除した手法。第一原理計算の中でも最も高精度な手法の一つである。
- DFT+U法
DFT計算において電子間の相関効果を補正するための近似手法。一電子近似による交換相関項の取り扱いに加え、局在電子に対して、一般ハバードモデルに倣ってオンサイトにおける量子力学的なクーロン相互作用を導入した手法。この手法において、有効オンサイトクーロン相互作用力は電子間の相関効果の程度を示す重要な変数となる。
- メタロセン分子
図1 (a)のような分子構造で、遷移金属元素を中心に、上下にシクロペンタジニル環(C 5 H 5 - )を配位子とする、D 5d 対称性のπ共役系分子。これまでに様々な金属元素を中心に配位させたメタロセン分子が報告されており、金属元素種に依存して電気的、磁気的性質が変化することから、分子スピントロニクス材料として有望な物質の一つとして知られる。