
CAPITAL: a major advance in single-cell RNA data analysis
Researchers from Osaka University have developed a computational tool called CAPITAL that can carry out accurate comparative analysis of complex single-cell sequencing datasets
New developments in high-throughput biological studies mean that the genes that are active in just a single cell can now be determined. However, analyzing the complex datasets that result can be challenging. Now, a team at Osaka University has developed CAPITAL, a new computational tool for comparing complex datasets from single cells.
RNA sequencing provides information on the subset of the entire population of genes that are actively being expressed, or are “switched on”. As technology has advanced, it has become possible to sequence the RNA population of just a single cell. This can provide a great deal of information on the specific changes in gene expression involved when a large population of mixed cells undergoes dynamic, transitional processes, such as differentiation or cell death, because each individual cell can be specifically analyzed rather than all the different cell types being pooled.
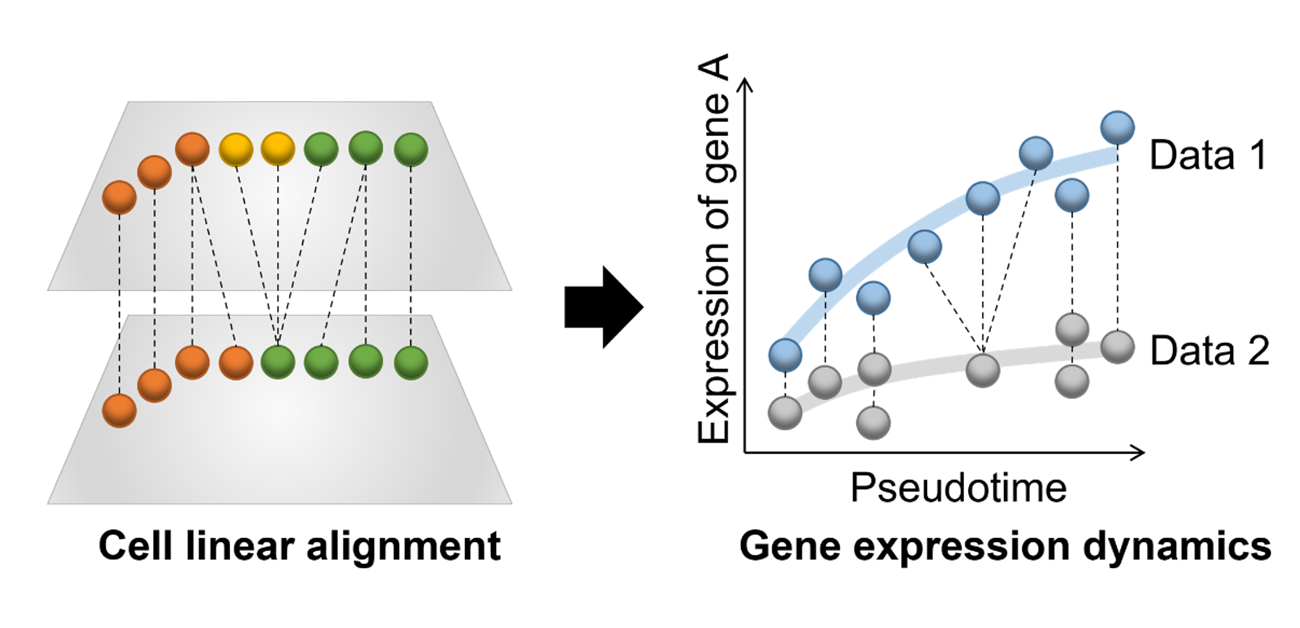
CAPITAL is specifically designed to compare complex datasets from single cells undergoing transitional processes. These analyses are carried out by defining a “pseudotime trajectory”, which places the cells along a hypothetical path that reflects their progress through the transitional process. These trajectories are not always straightforward and linear; they can become very complex and branching. In the past, only linear trajectories could be aligned for comparison, but the team’s innovation means that complex branching trajectories can now be accurately and automatically aligned and compared.
After developing the algorithm used for CAPITAL, which implements a method known as tree alignment, they tested it on both synthetic datasets and authentic datasets from bone marrow cells. The results demonstrated that CAPITAL is statistically more accurate and robust than the computational algorithms that existed previously, showing major advances over these methods.
Trajectory comparison is a powerful analysis that can, as an example, identify the gene expression dynamics between different species to provide information on evolutionary processes. “We showed in this study that CAPITAL can reveal the existence of different molecular patterns between humans and mice even when the expression patterns are similar and appear to be conserved,” says lead author Reiichi Sugihara. “This will allow the identification of novel regulators that determine cell fates.” This technology is not limited to just this type of data, as senior author Yuki Kato explains: “Our novel computational tool can be applied to a wide range of high-throughput datasets, including pseudotemporal, spatial, and epigenetic data.”
This powerful new technique will allow the global comparison of single-cell trajectories, which may lead to the identification of novel disease-associated genes that could not be identified by earlier comparative methods. Thus, CAPITAL represents a significant advance in the field of single-cell biology.
Fig. 1
Overview of CAPITAL: an algorithm for comparing pseudotime trajectories with branches
Credit: Yuki Kato
Fig. 2
CAPITAL is statistically better than data integration methods in trajectory conservation on 2,278 pairs of synthetic datasets with multiple branches
Credit: 2022 Sugihara et al., Alignment of single-cell trajectory trees with CAPITAL, Nature Communications
Fig. 3
Expression dynamics of gene CSF1 along pseudotime from hematopoietic stem cell to erythrocyte between human and mouse from bone marrow cells data
Credit: 2022 Sugihara et al., Alignment of single-cell trajectory trees with CAPITAL, Nature Communications
The article, “Alignment of single-cell trajectory trees with CAPITAL”, was published in Nature Communications at DOI: https://doi.org/10.1038/s41467-022-33681-3.