
Repair full of mistakes may develop cancer!?
Molecular mechanism of repair of DNA damaged by radiation clarified
A group of researchers led by Associate Professor SHINOHARA Miki at the Institute for Protein Research, Osaka University found that if DNA damage response (DDR) does not work when DNA is damaged by radiation, proteins which should be removed remain, and a loss of genetic information can be provoked, which, when repaired incorrectly, will lead to the tumor formation.
Radiation damages genomic DNA, the essential blueprint for life; therefore, living organisms have several mechanisms for maintaining the stability of their own genomes. Although they have big evolutionary differences, both humans and budding yeast contain proteins that perform the same function. This group examined the DNA repair function of yeast Xrs2, an orthlog of human Nbs1, and yeast Tel1, an orthlog of human ATM.
Mutations in the Nbs1 gene are responsible for a human hereditary disorder which develops a high risk of cancer. This group found that DNA damage was repaired when human hereditary disorder type mutations (xrs2 mutations) were introduced in yeast XRS2 genes, but it was repaired with more errors than a DNA sequence with no mutations. This group determined that the cause was that the function of the Tel1 protein, which is important for DNA damage response, was not fully exercised in xrs2 mutations.
In the process of DNA repair through homologous recombination, it is necessary to make double-stranded DNA near DNA lesions into single-stranded DNA. This group clarified that Ku remained on DNA damage in tel1 mutants and xrs2 mutants. Ku is not required, so it should be removed when DNA damage is repaired.
Ku is a protein to join DNA ends broken by non-homologous end joining (NHEJ). It is thought that Ku, a tool for repairing, joins DNA ends where Ku normally should not work, and as a result, repair is completed with incorrect DNA information.
Tumor formation occurs when genomic DNA is broken or errors have become continuous. In human cells as well, if Nbs1 and ATM function in the same way to ensure repair of DNA damage, tumor formation may be prevented.
This group’s achievement shows the possibility to clarify the mechanism of human tumor formation, especially the molecular mechanism of cell cancerization due to DNA damaged by radiation in the initial stage, by using the model of budding yeast, a primitive eukaryote. Furthermore, it may be possible to clarify the molecular mechanism of cancerization by radiation exposure through verification using human cells.
Author Summary
Genomic DNA provides the essential blueprint for life, and therefore living organisms have several mechanisms for maintaining the stability of their own genomes. DNA double-strand breaks (DSBs) are one of the most severe forms of DNA damage, which, without precise repair, can provoke a loss of genetic information, leading to tumor formation. DSBs are repaired by two distinct pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), which can be precise or imprecise. In addition, the DNA damage response (DDR) is essential in the cell to integrate multiple events that need to occur after damage: activation of DNA repair enzymes, selection of repair pathway and control of cell cycle progression, transcription, and so on. Here we show that different domains of Xrs2, a central DSB repair protein in budding yeast whose human ortholog, Nbs1, is linked to a human hereditary disorder with a high risk of cancer, is required not only for repair pathway choice but also for full activation of DDR. This result indicates that DSB repair and the DDR are coordinated at multiple levels to ensure precise repair and thus to maintain genomic integrity.
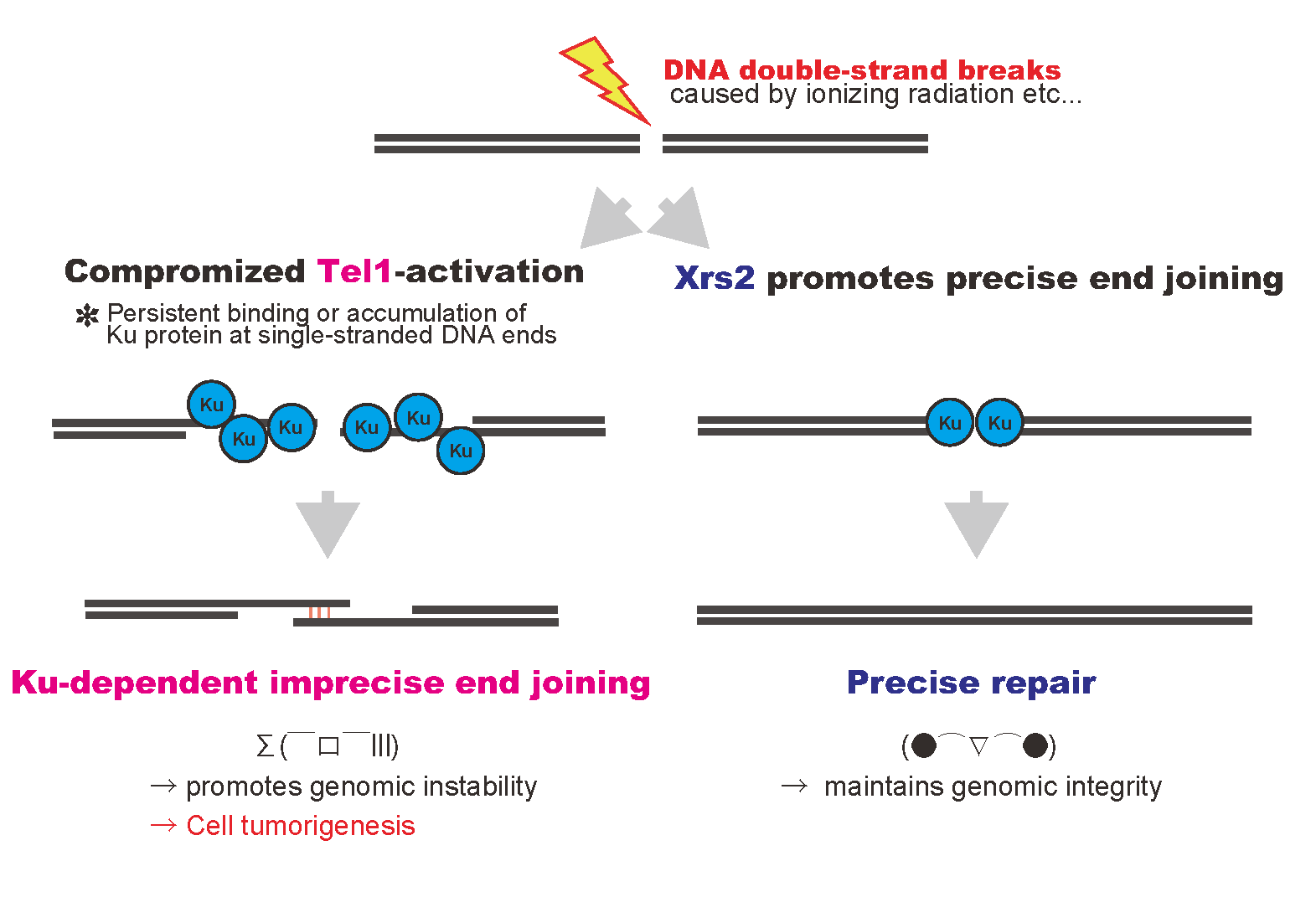
DNA double-strand breaks, introduced by ionizing radiation or others, are repaired rapidly and precisely in normal cells (right pathway). In contrast, compromised Tel1 activation with inefficient end joining activity in the mutant promotes accumulation of Ku protein at single-stranded DNA ends at damaged site, and then, results imprecise repair of genomic DNA (left pathway).
To learn more about this research, please view the full research report entitled " The MRX Complex Ensures NHEJ Fidelity through Multiple Pathways Including Xrs2-FHA–dependent Tel1 Activation " at this page of the PLOS Genetics website.
Related links