
Identified a novel therapeutic indicator for the designated intractable disease, immune thrombocytopenia (ITP)
Classification of refractory patients based on complement activation
- In immune thrombocytopenia (ITP), the research group has, for the first time in the world, identified a subset of patients in whom complement activation is involved, and elucidated a potential diagnostic method that enables their simple detection in clinical practice.
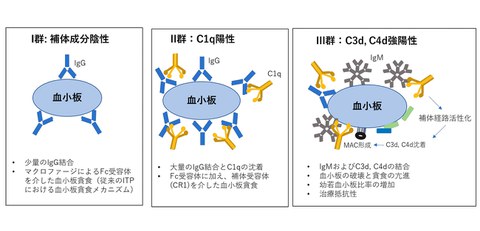
- Although the pathophysiology of ITP is heterogeneous and the causes and mechanisms of platelet destruction have not been fully clarified, analysis of molecular species bound to the surface of platelets in ITP patients showed three distinct mechanisms of platelet destruction.
- The result suggests the potential to easily diagnose patients who may benefit from complement inhibitors and is expected to guide better treatment selection for refractory ITP patients.
Outlines
A research group including Professor Naoki Hosen, together with Keiichi Nakata (graduate student) and Guest Professor Hirokazu Kashiwagi at the Immunology Frontier Research Center (WPI-IFReC), and the Department of Hematology and Oncology, Graduate School of Medicine, The University of Osaka, in collaboration with Ichio Onami of Chugai Pharmaceutical Co., Ltd. and Hiroaki Matsushita (affiliated with IFReC and the Graduate School of Medicine, The University of Osaka) has, for the first time worldwide, identified a subset of immune thrombocytopenia (ITP) patients in whom complement activation is involved, and elucidated a diagnostic method with the potential to detect this subset in clinical practice.
Immune thrombocytopenia (ITP) is an autoimmune disease characterized by a reduced platelet count caused by anti‑platelet autoantibodies, and it is designated as an intractable disease by the Ministry of Health, Labour and Welfare in Japan. In recent years, substantial progress has been made in the treatment of ITP; however, some cases remain refractory to conventional therapies and clarifying the mechanisms underlying thrombocytopenia in such refractory cases, as well as developing effective treatment strategies, has remained a major challenge.
In this study, the research group analyzed samples from 40 patients with ITP. By using flow cytometry, they demonstrated that patterns of complement deposition on the surface of platelets in ITP patients can be classified into three distinct groups. Particularly in Cluster 3, characterized by advanced complement activation, the study revealed that not only IgG autoantibodies but also IgM autoantibodies may be involved, that platelet destruction is pronounced and associated with an increased proportion of immature platelets, and that many cases show poor responses to conventional therapies.
Today, drugs targeting complement activation are being actively developed, and this approach is expected to help guide the selection of more effective treatments for refractory ITP patients.
Research Background
ITP is an autoimmune disease in which platelet counts are reduced due to the phagocytosis and destruction of platelets, primarily in the spleen, mediated by IgG autoantibodies bound to platelets. ITP is characterized by bleeding manifestations, such as subcutaneous hemorrhage (purpura) and impaired hemostasis, and was previously referred to as idiopathic thrombocytopenic purpura. ITP occurs in children, young women, and elderly people; however, particularly in adults, it often becomes chronic and requires long-term treatment, and is therefore designated as a intractable disease.
Treatment has traditionally included corticosteroids to suppress IgG autoantibody production, immunosuppressive agents, and splenectomy. In recent years, however, significant progress has been made with the demonstrated efficacy of thrombopoietin receptor agonists, which stimulate platelet production. Nevertheless, a certain proportion of patients remain refractory to these treatments, and elucidating the mechanisms underlying thrombocytopenia in such cases, as well as developing effective therapeutic strategies, has remained a major challenge.
Research Contents
The research group collected blood samples from 40 ITP patients and quantified complement components (C1q, C3d, and C4d) as well as IgG and IgM antibodies bound to the platelet surface using flow cytometry. IgM antibodies were predominantly detected in Cluster 3, whereas IgG antibodies were observed in both Cluster 2 and Cluster 3. Furthermore, cases refractory to first-line corticosteroid therapy were observed more frequently in Cluster 3 than in Cluster 1.
In ITP, enhanced platelet destruction leads to a shortened platelet lifespan, resulting in an increased proportion of young platelets (immature platelet fraction) which can be measured easily and rapidly using automated hematology analyzers. Furthermore, complement deposition on platelets was found to correlate with an increased immature platelet fraction, suggesting that enhanced platelet destruction associated with complement activation contributes to thrombocytopenia, particularly in Cluster 3, and may be linked to resistance to conventional therapies.
Social Impacts
This study demonstrated that there exists a subset of patients with ITP in whom complement activation plays a significant role and may be associated with resistance to treatment and further suggests that such patients may be identified using a simple method, namely measurement of the immature platelet fraction. In recent years, the development of new therapeutic agents for refractory ITP has advanced, including complement inhibitors. This study is expected to help guide the selection of more effective treatments for patients with refractory ITP.
Notes
The article, “Complement activation profile in adult primary immune thrombocytopenia,” was published in British Journal of Scientific Reports at DOI: https://doi.org/10.1182/blood.2025032255