
Scientists clarify how genetic mutations cause abnormalities in brain development and sociality
Identifying underlying mechanisms will lead to the development of potential autism spectrum disorder drugs
A group of researchers from Osaka University and the National Center of Neurology and Psychiatry has found that POGZ (Pogo transposable element derived with ZNF domain) gene is necessary for the development of a healthy brain and that de novo mutations in POGZ impair neuronal development in mouse brains and cause autism spectrum disorder (ASD)-related behavioral abnormalities.
POGZ has been identified as a recurrent de novo mutated gene in patients with neurodevelopmental disorders (NDDs), including ASD. A spontaneous de novo mutation is a new mutation that manifests in a child that neither the parent carries, meaning the mutation is not inherited. It is suggested that mutation in POGZ is a risk factor for developing ASD; however, the role of POGZ in brain development and the biological significance of ASD-associated de novo POGZ mutations in the etiology of ASD are unknown.
In this study, during embryonic neurogenesis from embryonic day 14.5 (E14.5) to E18.5, POGZ was highly expressed in the cortical neural stem cells (NSCs) and intermediate progenitor cells (IPs) in the ventricular and subventricular zones. In a 14-day-old mouse embryo, the neuronal migration was significantly inhibited by POGZ knockdown. POGZ was likely to regulate transcriptional networks that control neuronal differentiation and play an important role in cortical neuronal development. POGZ mutations impaired POGZ function in neuronal development.
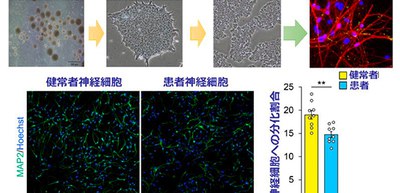
The researchers established induced pluripotent stem cell (iPSC) lines using immortalized B cells obtained from an ASD patient carrying the Q1042R mutation of POGZ and an unaffected healthy control, differentiating each iPSC lines into NSCs. The proportion of differentiated neurons was significantly lower in the patient-derived NSCs than in the control NSCs, suggesting that neuronal differentiation was impaired in the patient-derived NSCs.
This group developed the first mouse model that carried a pathogenic de novo mutation of POGZ identified in an ASD patient, POGZWT/Q1038R mice (wild-type Q1038R-mutated POGZ mice). (Q1038R mutation in mouse POGZ corresponds to the Q1042R mutation in human POGZ.)
A reciprocal social interaction test revealed that (a) juvenile POGZWT/Q1038R mice spent less time in sniffing novel mice than their littermates did, suggesting impaired social interaction in juvenile POGZWT/Q1038R mice and (b) after separation from their mothers, POGZWT/Q1038R pups emitted more and longer ultrasonic vocalization (USV) calls than their wild-type littermates did, suggesting a disturbance in communications from POGZWT/Q1038R pups to their mothers.
Since the excitatory neurons in the cerebral cortex were hyperactivated in POGZWT/Q1038R, the researchers thought that pharmacological inhibition of synaptic transmission could rescue the impaired social interaction typical of POGZWT/Q1038R.
Results of this study suggest that a de novo mutation could be one of the causes of ASD. The results revealed that an altered cellular balance of excitation and inhibition within neural circuitry might cause the social and cognitive deficits and that these abnormalities were pharmacologically treatable even in adulthood. This group’s achievements will lead to the development of ASD drugs that target neuronal development regulated by POGZ..
The article, “Pathogenic POGZ mutation causes impaired cortical development and reversible autism-like phenotypes” was published in Nature Communications at DOI: https://doi.org/10.1038/s41467-020-14697-z.
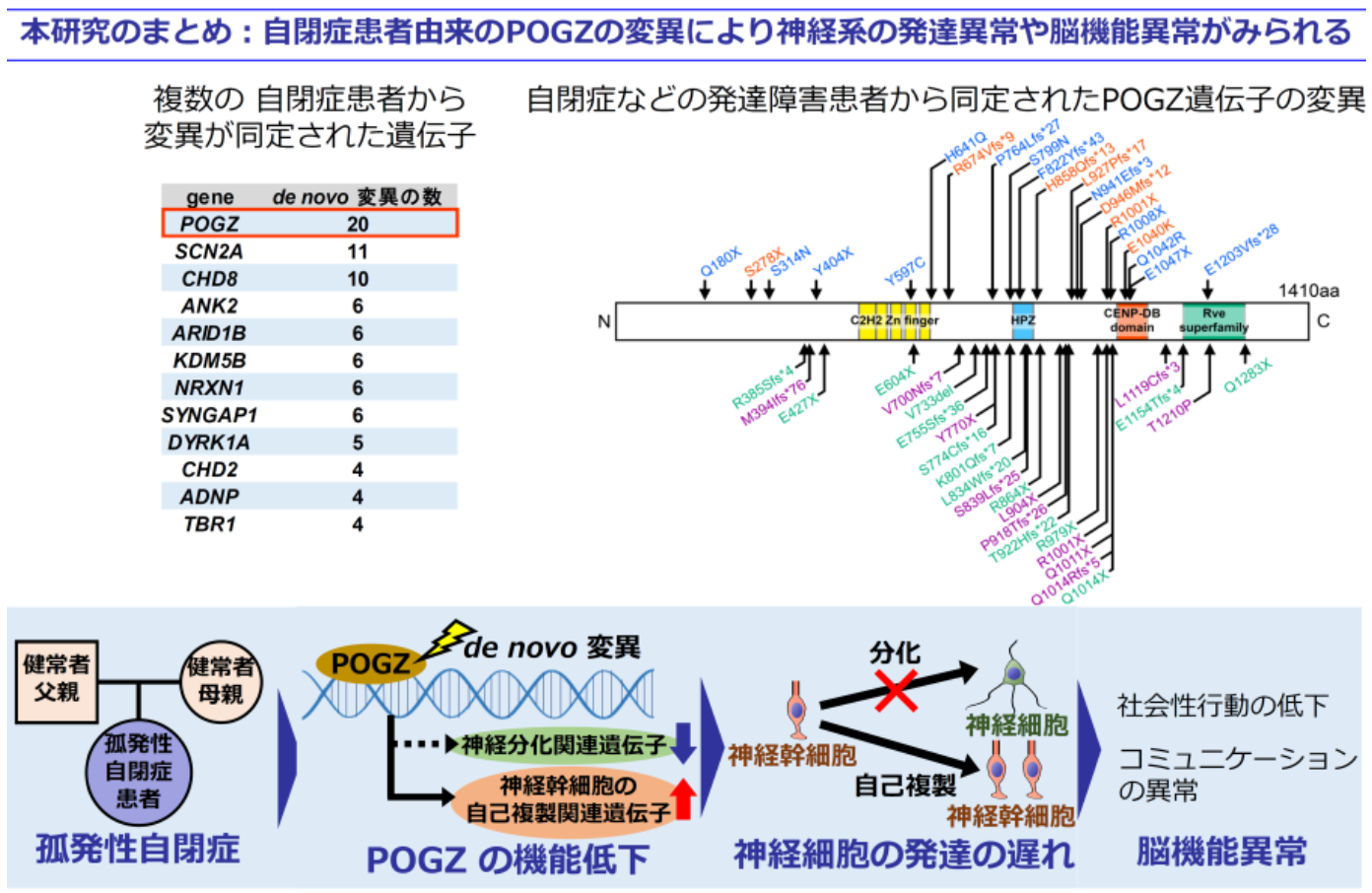
Figure 1
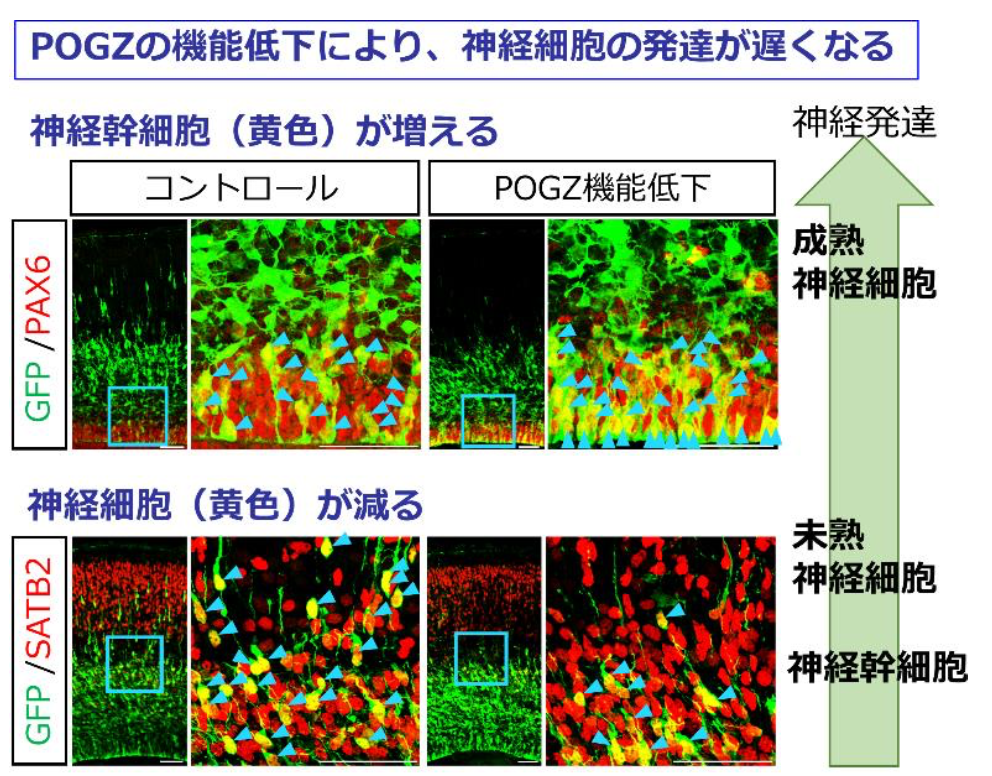
Figure 2
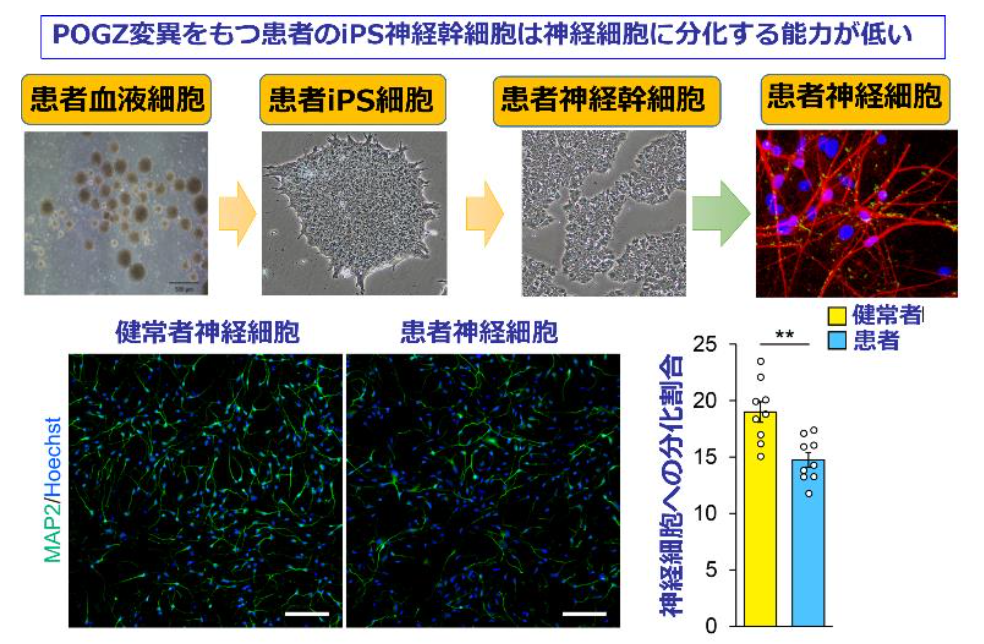
Figure 3
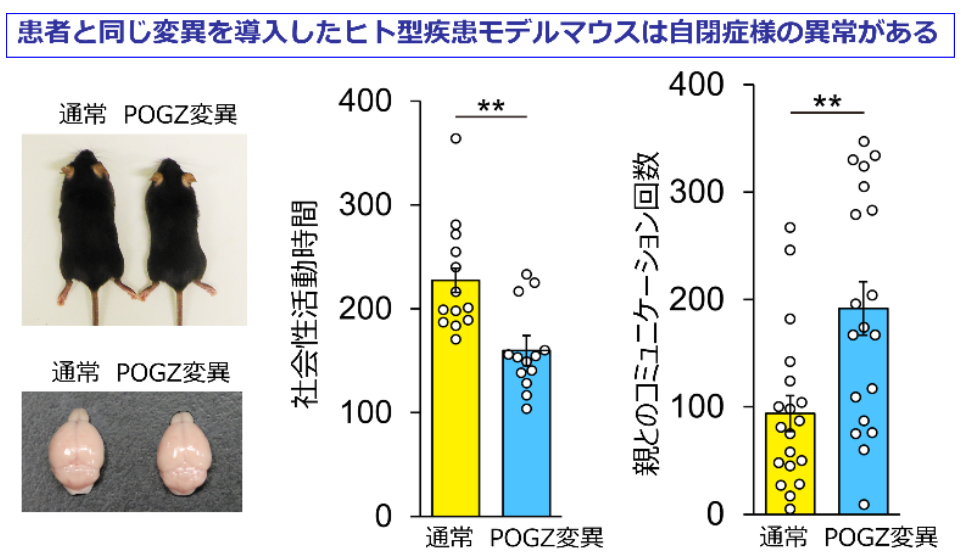
Figure 4
Related links
Graduate School of Dentistry, Osaka University (link in Japanese)