
Impaired glycolysis: A factor may lead to DiGeorge/del22q11 syndrome
A group of researchers from Osaka University, University of Chicago (U.S.), RIKEN, Keio University, The University of Tokyo, and the Tokyo Institute of Technology has clarified the possibility that impaired glycolysis is related to DiGeorge/del22q11 syndrome.
DiGeorge syndrome (DGS), also known as 22q11.2 deletion syndrome, is caused by the heterozygous deletion of a portion of chromosome 22 at position q.11 (del22q11). This syndrome includes serious congenital disorders, such as cardiac diseases, immune deficiency, and urological diseases, but the mechanisms that underlie DGS and other related congenital syndromes were unknown and there were no clues regarding their treatment.
In order to elucidate the mechanisms behind DGS and other related congenital syndromes, the researchers investigated mouse models in which CRK, CRKL, or both could be disrupted conditionally. Developmental defects in the mouse models had similarities to DGS, and normal development of the affected tissues was sensitive to the combined gene dosage of the CRK (CT10 regulator of kinase) gene and CRKL (CRK Like) proto-oncogene.
CRK and CRKL encode proteins. These proteins function as adaptor proteins in tyrosine kinase, which are important mediators of a signal transduction process, determining key roles in diverse biological processes like growth, differentiation, metabolism and apoptosis in response to external and internal stimuli. CRKL is tightly linked to cancer and induces cancer cell proliferation and invasion.
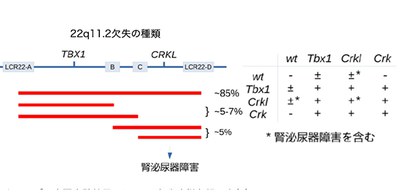
This group clarified that a deletion of candidate causative genes, namely the CRK family genes (CRK and CRKL) and TBX1, caused serious morphological defects such as ventricular septal defects in mice. CRK and CRKL, two paralogs (the two genes that exist after a gene duplication event) are localized to 17p13.3 and 22q11.21 in the human genome, respectively. CRK is not a 22q11 gene, but homozygous deficiency of either CRK or CRKL and a heterozygous deletion in CRK/CRKL in mice caused morphological defects, which resembled DiGeorge anomaly. This suggested that the shared functions of CRK and CRKL were important in the fetal development process in which DiGeorge anomaly is caused.
Using cells from knockout mice, the researchers investigated common functions of CRK and CRKL, finding that several metabolites in the central glucose metabolism pathway, glycolysis, were decreased. CRK/CRKL deficiency sensitized Mouse Embryonic Fibroblast (MEFs) to 2-deoxy-D-glucose, a competitive inhibitor of glycolysis, to induce cell blebbing, which might be an indication of apoptosis. In addition, activated Rapgef1, a CRK/CRKL-downstream effector, restored expression of some glycolysis enzyme genes that were down-regulated in CRK/CRKL deficiency. CRK/CRKL-shared pathways orchestrated metabolic homeostasis and cell behavior.
From these findings, they speculated that glucose metabolism might be a possible contributing factor that could partly explain large variations of penetrance and expressivity observed among DiGeorge/22q11.2DS patients.
Their achievements will lead to the development of new treatment methods for DiGeorge/del22q11 syndrome by improving glucose metabolism control through nutritional regulation during fetal life.
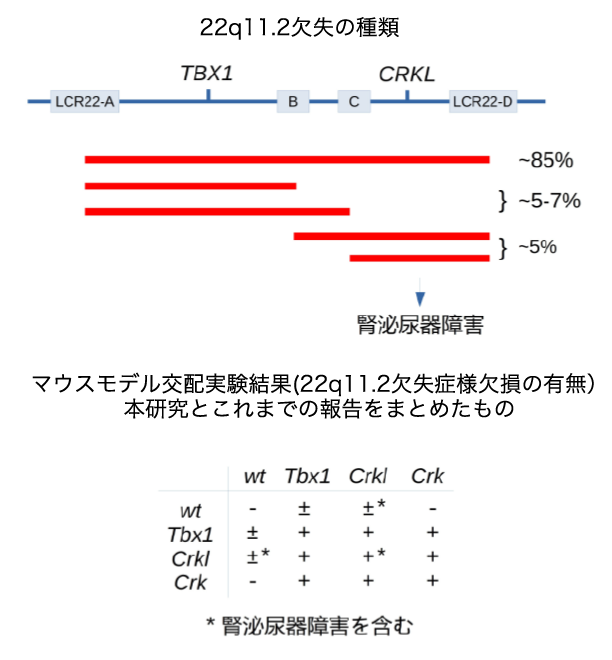
Figure 1
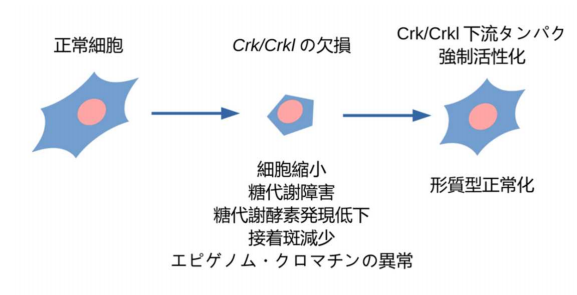
Figure 2
Figure 3
The article, “Essential role of the Crk family-dosage in DiGeorge-like anomaly and metabolic homeostasis ,” was published in Life Science Alliance at DOI: https://doi.org/10.26508/lsa.201900635.
Related links