
Faulty DNA repair depresses neural development
Osaka University researchers discover DNA polymerase β (Polβ) deficiency in neural stem cells affects neuronal survival and neural network in the developing brain.
DNA is the computer code that programs every event in the body. Despite the importance of DNA fidelity, as the body develops, cells grow and replicate, DNA is constantly turned over. This repeated process can compromise the DNA, which is why the body has many DNA repair machineries. Using mice, Osaka University scientists report a defect in one type of machinery, DNA polymerase β (Polβ), causes underdevelopment of the brain’s cortices and axonal network. The findings could help explain cortical development disorders, such as autism and microcephaly.
“Polβ is responsible for repairing DNA base damage in the brain. Because many neurological disorders are associated with de novo mutations, we wanted to study how loss of Polβ affects neuronal development,” said Assistant Professor Noriyuki Sugo, an expert in the study of Polβ in brain development.
“We found evidence that Polβ has a role in the development of the brain but not other organs and that its defect causes catastrophic DNA double strand breaks (DSBs), and consequent cell death in certain regions of the developing cortex,” he continued.
These regions represent one of the earliest stages of cortical development, and the generation of cortical neurons is fundamental for proper neural networking.
In the present study, Sugo and his team prepared mutant mice deficient in Polβ. These mice showed a large number of DSBs in neural progenitors, the stem cells that eventually produce neurons. Consequently, many immature neurons went on to die through apoptosis. Furthermore, the mice showed defects in the development of specific brain anatomy and the growth of axon in specific cell types, suggesting both an underdevelopment of the cortex and of neural networking.
“We found that Polβ deficiency led to higher neuronal cell death in deeper layers than upper layers of the cortex. The deeper layers were thinner,” said Sugo. He added that deeper-layer neurons were marked by a higher rate of DSBs.
Neurons formed in these layers are thought essential to the early stages of neural networking. Thus, even if the cells manage to escape death, the brain circuitry is likely compromised.
Finally, proper development depends on both genetic and epigenetic factors. The correction of DNA damage by Polβ is an example of genetic regulation. In addition, the researchers found DNA demethylation, an example of epigenetic regulation, is also abnormal in mice deficient of Polβ. Together, Sugo argues the findings are strong evidence for the importance of Polβ on proper gene expression in cortical development and provide a new target for the study of associated syndromes and disorders.
“The brain is actively constructed in embryonic stages. Neural progenitors produce many neurons, their genomic DNA is constantly processed. Defects in Polβ function could be a new target for explaining cortical developmental disorders.”
Abstract
DNA repair is crucial for genome stability in the developing cortex, as somatic de novo mutations cause neurological disorders. However, how DNA repair contributes to neuronal development is largely unknown. To address this issue, we studied the spatiotemporal roles of DNA polymerase β (Polβ), a key enzyme in DNA base excision repair (BER) pathway, in the developing cortex using distinct forebrain-specific conditional knockout mice, Emx1-Cre/Polβ fl/fl and Nex-Cre/Polβ fl/fl mice. Polβ expression was absent in both neural progenitors and postmitotic neurons in Emx1-Cre/Polβ fl/fl mice, while only postmitotic neurons lacked Polβ expression in Nex-Cre/Polβ fl/fl mice. We found that DNA double-strand breaks (DSBs) were frequently detected during replication in cortical progenitors of Emx1-Cre/Polβ fl/fl mice. Increased DSBs remained in postmitotic cells, which resulted in p53-mediated neuronal apoptosis. This neuronal apoptosis caused thinning of the cortical plate, although laminar structure was normal. In addition, accumulated DSBs also affected growth of coticofugal axons but not commissural axons. These phenotypes were not observed in Nex-Cre/Polβ fl/fl mice. Moreover, cultured Polβ-deficient neural progenitors exhibited higher sensitivity to the base-damaging agent methylmethanesulfonate, resulting in enhanced DSB formation. Similar damage was found by vitamin C treatment, which induces TET1-mediated DNA demethylation via 5-hydroxymethylcytosine. Taken together, genome stability mediated by Polβ-dependent BER is crucial for the competence of neural progenitors, thereby contributing to neuronal differentiation in cortical development.
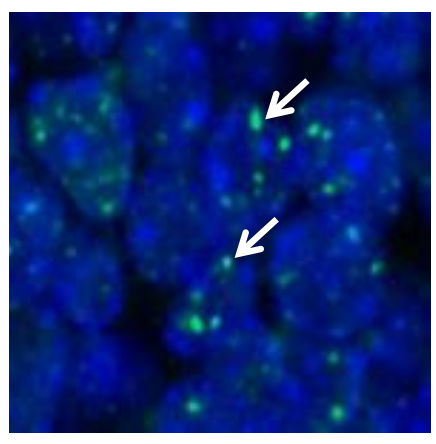
Figure 1. Increased DNA double-strand breaks in Polb-deficient neural progenitors.
To learn more about this research, please view the full research report entitled " Genome Stability by DNA polymerase β in Neural Progenitors Contributes to NeuronalDifferentiation in Cortical Development " at this page of the Journal of Neuroscience website.
Related link