
キンギョの全ゲノム解読により脊椎動物の進化の謎に迫る
新型次世代 DNA シーケンサーを使って全ゲノム重複と進化の謎解明に道筋
研究成果のポイント
・キンギョの全ゲノム 配列の解読に世界で初めて成功し、1400万年前に全ゲノム重複 が起こったことや、この間に重複した遺伝子が淘汰や進化を遂げる様子が明らかとなった。
・キンギョは一般の魚類の約2倍の遺伝子を持ちゲノム構造が複雑で全ゲノム解読が困難であったが、新型のロングリード次世代DNAシーケンサー と雌性発生 キンギョを用いたことで解読が可能となった。
・ヒトを含む脊椎動物の進化や「体の形づくり」の仕組みの解明、新たなキンギョ品種の作出、ヒト疾患の発症機構の解明への貢献が期待される。
概要
大阪大学蛋白質研究所・分子発生学研究室(古川貴久教授)の大森義裕招へい教授(当時准教授。現長浜バイオ大学教授)らの研究グループは、国立遺伝学研究所・川上浩一教授、藤山秋佐夫特任教授、愛知県水産試験場内水面漁業研究所弥富指導所、および米国国立衛生研究所(NIH)Shawn Bargess上席研究員らと共同でキンギョの全ゲノム配列を世界で初めて解読し、キンギョの祖先種(フナの仲間)のゲノムが約1400万年前に倍加したことや(全ゲノム重複)、倍加した遺伝子群の一部が進化の過程で淘汰されたり、新たな発現パターンを獲得したりする様子を明らかにしました(図1)。今回の研究では、新型のロングリード次世代DNAシーケンサーを使ったことと、通常の交配から得たキンギョではなく、ゲノム構造がシンプルな雌性発生キンギョを用いたことがブレイクスルーに繋がりました。キンギョは約千年前にフナから選別・育種され、現在、デメキンやランチュウ、リュウキンなど数十種類の多様な品種が飼育されています。キンギョは、その形態的な多様性から、私たち、人間を含む脊椎動物の体の形を決めるメカニズムの解明に役立つと考えられていますが、全ゲノムが解読されていないため遺伝子レベルでの研究が困難でした。今回のゲノム解読により、体の形を決めるメカニズムや全ゲノム重複と進化の研究を進めるための新たな扉が開かれました。また、キンギョの品種にはヒトの病気と似た症状をもつものがあり、キンギョがヒトの病気の原因解明や診断・治療法の確立に役立つ可能性も期待されることから、アメリカなど海外でもそのゲノム情報の研究に注目が集まっています。
本研究成果は、米国科学誌「Science Advances」に、6月27日(木)午前3時(日本時間)に公開されました。
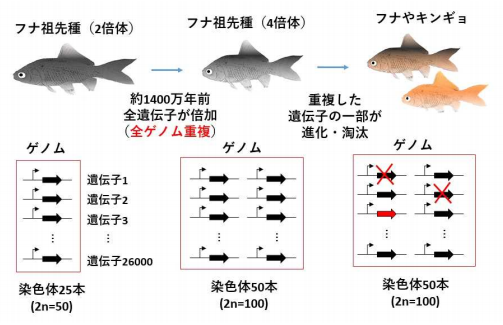
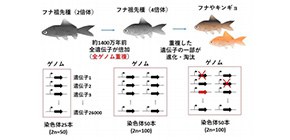
図1 キンギョとフナの祖先種での全ゲノム重複と進化
キンギョやフナの祖先種では、約1400万年前に全遺伝子が倍加する現象(全ゲノム重複)が起こり、遺伝子が一般の魚類の2倍となった。その後、倍加した遺伝子の一部は進化・淘汰されている。その変遷の様子が今回のゲノム解読により明らかとなった。
研究の背景と結果
コイ科の魚であるキンギョは約千年前の中国、宋の時代に野生のフナから育種が始まり、主に東アジアにおいて数百年間にわたって品種改良が進められてきました。日本には室町時代に伝来し、江戸時代に盛んに品種改良がおこなわれ、現在、愛知県弥富や奈良県大和郡山、東京都江戸川などの産地を中心に育種が継承されています。デメキンやランチュウ、オランダ、シュブンキンなど数十種類の多様な品種が飼育されています。キンギョの体の形の多様性は19世紀のチャールズ・ダーウィンの時代に既に科学的な関心の対象となっており、彼の著書の中でもその多様性についての興味がつづられています。
また、キンギョは私たちヒトを含む脊椎動物の仲間であり、その体の形づくりのメカニズムは共通する部分が多く、それに必要な遺伝子も共通していると考えられています。すなわち、キンギョの体の形づくりのメカニズムを研究することで、ヒトを含む脊椎動物の体の形づくりのメカニズムを解明できると期待されます。それはヒト疾患発症のメカニズムの理解にもつながります。このように、キンギョは研究の対象として興味が持たれていたのですが、多様性や体の形づくりに関わる遺伝子群の全貌を明らかにするためには、全ゲノム塩基配列情報が必要です。けれども、キンギョの染色体では異質4倍体化(全ゲノム重複)がおこっており、一般の魚類の約2倍の遺伝子を持つことから、ゲノム構造が複雑で全ゲノム解読はこれまで成功していませんでした。私たちは、今回、この問題を克服するために新型のロングリード次世代DNAシーケンサーを用いました (図2) 。これまでのショートリード次世代DNAシーケンサーでは、100から200塩基程度の短いDNA配列を大量に解析し、それらをパズルを解くように繋ぎ合わせることで、全長18億塩基対に及ぶ全ゲノムの解読を進めることになります。このため配列不明の部分(ギャップ)が発生しやすいという欠点がありました。今回用いた新型のロングリード次世代DNAシーケンサーでは旧型の100倍以上の長さのDNA塩基配列(1万から4万塩基)を一気に読み進めることができるのでより正確なゲノム情報にもとづいて遺伝子地図を作製することができます。全ゲノム重複が起こっている複雑なキンギョゲノムを読み解くにはロングリード次世代DNAシーケンサーによる解読が威力を発揮しました。
もう一つの工夫は雌性発生キンギョを用いたことです (図3) 。通常のキンギョは私たちヒトと同様に父母両方の対立遺伝子を受け継ぐのですが、キンギョの発生段階で特殊な処理をすることにより、母親由来の染色体からなるキンギョ(雌性発生キンギョ)を作製しました。このキンギョでは多くの遺伝子が母親由来の遺伝子のみとなっています。また、今回の研究では野生のフナに近い体形を持つワキンというキンギョ品種を用いました。このことによりゲノム構造をシンプルにし、解読を容易にすることに成功しました。これらの手法により、キンギョの祖先種のゲノムが約1400万年前に倍加(全ゲノム重複)したことや、倍加した遺伝子群の一部が進化の過程で淘汰されたり、新たな発現パターンを獲得したりする様子が明らかとなりました。
全遺伝子数が倍加する全ゲノム重複は、私たちヒトを含む脊椎動物の進化に重大な影響を与えたと考えられていますが、約5億年前に起こったと考えられる現象なので、その後、重複した遺伝子がどのように進化して現在の私たち脊椎動物のゲノムを作り上げているかはよくわかっていません。キンギョのゲノム研究をすすめることで、全ゲノム重複後の遺伝子進化のメカニズムを明らかにする手掛かりが得られると考えられます。
今回の研究で、キンギョの祖先種で1400万年前に全ゲノム重複が起こったことや、その後、倍加した遺伝子の12%が淘汰されて無くなっていること、また、重複している遺伝子の約30%が臓器で新たに発現するようになるなどの進化を遂げたことがわかりました。また、遺伝子の淘汰や発現変化を受けやすい遺伝子の顔ぶれがより鮮明になってきました。遺伝子が失われる速度は、8000万年前に全ゲノム重複が起こったサケと比べて1.7倍速く、キンギョのゲノムが急速に遺伝子を失う進化過程にあることが明らかとなりました。
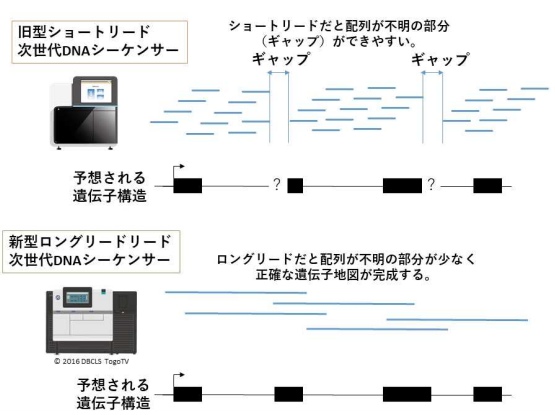
図2 ロングリード次世代DNAシーケンサーによるゲノム解読
これまでのショートリード次世代DNAシーケンサーでは、100から200塩基程度の短いDNA配列を大量に読み繋ぎ合わせることで、18億塩基対に及ぶ全ゲノムの解読を進めるため、配列不明の部分(ギャップ)ができやすかった。今回用いた新型のロングリード次世代DNAシーケンサーでは1万から4万塩基を一気に読み進めるので、より正確な遺伝子地図を作製することができる。全ゲノム重複が起こっている複雑なキンギョゲノムを読み解くにはロングリード次世代DNAシーケンサーによる解読が威力を発揮した。
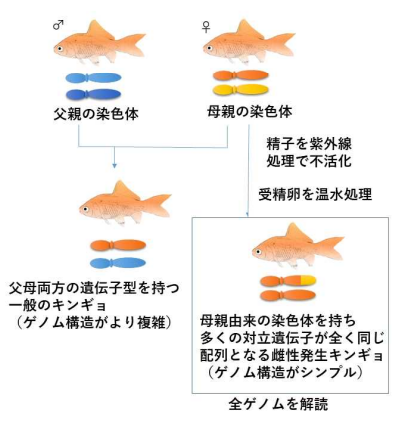
図3 雌性発生キンギョの作出と染色体
一般のキンギョは父親由来と母親由来の染色体を持つ。これらは、微妙に配列が異なるため、ゲノム構造が複雑となり解読の障害となる。一方、今回用いた雌性発生キンギョは母親由来の染色体のみを持ち、多くの対立遺伝子が全く同じ配列となっているのでゲノム構造がシンプルで解析が容易となった。
本研究成果が社会に与える影響(本研究成果の意義)
本研究成果により、今後、キンギョ全ゲノム配列情報を使って進化の研究や、脊椎動物のかたちや色をつくるメカニズムの研究が加速することが予想されます。キンギョは、私たちヒトを含む脊椎動物の多様性や体づくりのメカニズムについての研究を進める上で興味深い対象なのですが、これまで全ゲノムが解読されていなかったため、遺伝子レベルでの正確な解析が困難でした。私たちは、今回のゲノム解読により、これらのメカニズムの解明へ向けて扉が開かれたと考えています。また、キンギョの品種にはヒトの病気と似た症状をもつものがあり、キンギョがヒトの病気の原因解明や診断・治療法の確立に役立つ可能性も期待されます。
現在、私たちは今回得られた全ゲノム配列のデータをもとに、デメキンやランチュウを含む様々なキンギョ品種のゲノム解析を行うことで脊椎動物の体の形をつくる遺伝子の探索をすすめています。また、新たなキンギョ品種の作出や病原菌に対する耐性などキンギョの品種改良にも貢献することが期待されます。
特記事項
本研究成果は、2019年6月27日(木)午前3時(日本時間)に米国科学誌「Science Advances」(オンライン)に掲載されました。
タイトル:“De Novo assembly of the goldfish (Carassius auratus) genome and the evolution of genes afterwhole genome duplication”
著者名:Zelin Chen*, Yoshihiro Omori*, Sergey Koren, Takuya Shirokiya, Takuo Kuroda, Atsushi Miyamoto, Hironori Wada, Asao Fujiyama, Atsushi Toyoda, Suiyuan Zhang, Tyra G. Wolfsberg, Koichi Kawakami, Adam M. Phillippy, NISC Comparative Sequencing Program, James C. Mullikin, and Shawn M. Burgess
* equally contribution
参考URL
大阪大学 蛋白質研究所 蛋白質高次機能学研究部門 分子発生学研究室
http://www.protein.osaka-u.ac.jp/furukawa_lab/index.html
用語説明
- ゲノム
ひとつの生物の染色体1セットに含まれているすべての遺伝情報。例えばヒトのゲノムは約30億塩基対から成り約2万個の遺伝子を持っている。
- 全ゲノム重複
生物のゲノム全体が倍化する現象。同種のゲノムのコピーが倍加する同質倍数体と、進化的に近い異種の交雑によって起こる異質倍数体がある。動物や植物、菌類でも見られる。本来、全遺伝子数が数千から数万個の生物種の遺伝子数が倍加することで、適応の自由度が増し生物の多様性が生み出されると考えられている。倍加した遺伝子数は数千万年オーダーの時間をかけて徐々に減少し、もとの遺伝子数に戻ってゆく。この過程で重複した遺伝子の一部は機能獲得や機能分化することで進化する。脊椎動物では、過去に2回の全ゲノム重複を経験したと考えられており、これらの全ゲノム重複後の遺伝子進化が脊椎動物の進化の大きな原動力となっていると考えられている。
- ロングリード次世代DNAシーケンサー
これまで主流であった、ショートリード次世代DNAシーケンサー(第2世代DNAシーケンサー;イルミナ社HiSeqシリーズなど)では、一度に100塩基から200塩基のDNA配列を大量に解析することで、ゲノム配列解析を行っていた。これに対し、新型のロングリード次世代DNAシーケンサー(第3世代DNAシーケンサー;PacificBiosciences社PacBio RS II など)では、一度に1万から4万塩基のDNA配列を解析できるので、ゲノムサイズの大きな生物のより正確なゲノム解析が可能である。
- 雌性発生
脊椎動物では、精子に含まれる父親由来の染色体と卵子に含まれる母親由来の染色体が、受精により合わさって一つの個体として発生する。一方で、紫外線などで父親由来の染色体を不活化した精子を用いて、受精させ、受精後に温度や圧力などの外的環境を変化させることで、母親由来の染色体のみから成る個体を得ることができる。こうして得られた個体が雌性発生個体であり、ゲノム配列の多様性が少ないため、ゲノム解析が容易となる。