

In situ click chemistry: Having target proteins assemble their own inhibitors
KDM5C inhibitor, a candidate therapeutic agent for neurological disorders, identified
A group of researchers from Osaka University, Kyoto Prefectural University of Medicine, and Kyoto University has developed a screening method to identify ligands as novel drug candidates by having target proteins assemble their own inhibitors.
By applying the yield of the triazole products obtained in a metal ion-catalyzed in situ azide–alkyne cycloaddition (AAC) reaction to activity-based high-throughput screening, this group identified a potent and selective inhibitor of Fe(II)-dependent lysine demethylase 5C (KDM5C) in a short time. This inhibitor is a novel drug candidate and KDM5C is a metalloprotein, which causes cancer and neurological disorders and is a candidate therapeutic target gene in cancers and neurological disorders.
Various approaches have been developed to explore promising drug candidates. In order to reduce financial and time burdens in these approaches, techniques such as high-throughput screening (HTS) have been used in traditional drug development. However, activity-based HTS is not sufficiently sensitive to detect even potent ligands and hit compounds are not always highly active toward the target proteins.
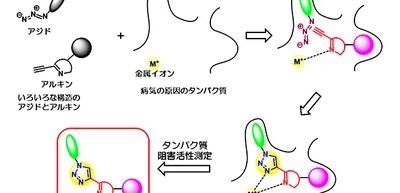
Thus, this group developed a method to identify inhibitors of metalloproteins by metal ion-catalyzed in situ click chemistry and activity-based assay. In situ click chemistry is a target-guided synthesis approach in which the target protein is engaged in assembly of its own ligand by AAC reaction.
In this study, focusing on a metalloprotein, the researchers discovered an inhibitor of KDM5C, which contains an Fe(II) ion in its active site, by employing the combination of metal ion-catalyzed in situ click chemistry and an activity-based high-throughput assay.
They demonstrated their method for screening drug candidates as follows:
1. They linked alkyne fragments (2-ethynyl N-heterocompounds), which can be coordinated on a metal ion (catalytic Fe(II) ion), with structurally diverse azides on a target protein KDM5C.
2. Alkyne fragments coordinated on the metal ion Fe(II) on KDM5C and azide fragments, which selectively bind in the pocket of enzyme, reacted effectively and produced triazoles via AAC reactions.
3. Triazoles strongly inhibited KDM5C due to the binary coordination, which contributed to an increase in the affinity of the triazole products and helped the researchers identify active ligands.
4. They assessed triazole activity using an activity-based assay, identifying a reaction product that shows potent and selective inhibitory activity toward KDM5C.
Since the combination of azide fragments and alkynes that cannot bind in a binding pocket of KDM5C does not produce the triazole products, the combination cannot become a drug candidate.
This method allows chemists to assess multiple combinations of alkyne and azide for exploration of drug candidates with less effort in a short period of time. This metal-catalyzed in situ AAC should be generally applicable to other metalloproteins.
Using this method, this group identified the inhibitor, a therapeutic drug candidate compound for treating neurological disorders, within only two days, although it would take more than 6 months using conventional methods of synthesizing compounds. (Patent applied: 2019-106166)
This method will make the development of innovative drugs faster and less costly.
Figure 1
The article, “Metalloprotein-Catalyzed Click Reaction for In Situ Generation of a Potent Inhibitor,” was published in Catalysis at DOI: https://pubs.acs.org/doi/full/10.1021/acscatal.0c00369.