
Identification of a significant modifier of the amount of amyloid-ß (Aß) peptide, strongly implicated in Alzheimer's disease, that accumulates in the brain of Alzheimer's patients.
Similar transcriptome analyses will certainly be applicable in identification of genetic factors in multifactorial disorders
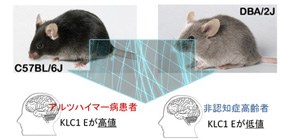
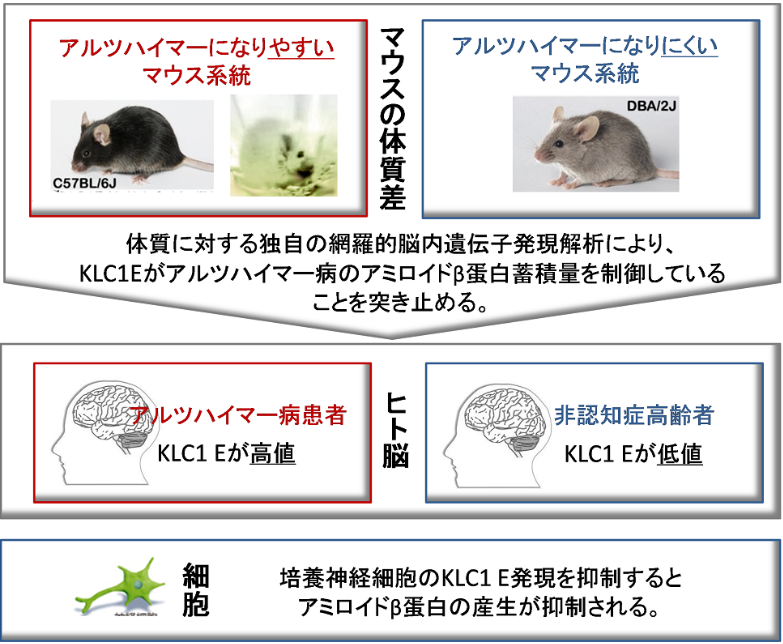
Under the leadership of MORIHARA Takashi (Professor, Course of Integrated Medicine, Graduate School of Medicine, Osaka University) a group of researchers identified, following genome-wide transcriptome analysis, that the kinesin light chain-1 (KLC1) splice variant E was a significant modifier of the amount of amyloid-ß (Aß) peptide, strongly implicated in Alzheimer's disease (AD), that accumulated in the brain.
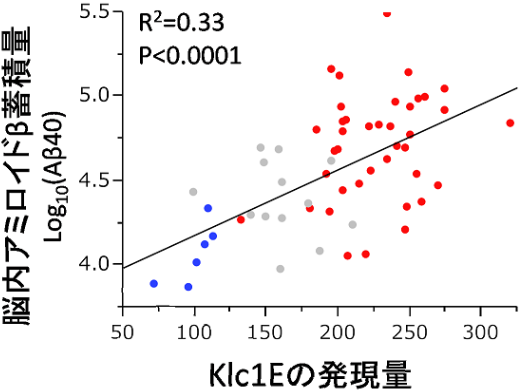
In general, genetic factors affect the incidence of diseases although there are constitution differences not only in humans, but also in mice. This group discovered that the tendency to develop AD differed by mouse strain; mice of certain strains developed AD more than those in other strains did. This indicates that genetic background plays a role to some degree in the development of AD. In order to identify suppressor genes of AD, this group conducted transcriptional analyses on different strains of mice. As a result, they identified a strong link between the amount of KLC1 and the amount of Aß accumulation. Additionally, they confirmed increased KLC1 splice variant E levels in AD patients' brains. Furthermore, when KLC1 splice variant E in neuroblastoma cells was artificially reduced, the production of Aß was suppressed.
This group's discovery suggests that the dysfunction of intercellular trafficking is associated with the development of AD and will lead to the development of epoch-making diagnosis and therapeutic methods. The clarification of human diseases through this group's comprehensive analyses of gene expression based on genetic differences in mouse strains will be used in the identification of genetic factors of multifactorial disorders including AD.
Abstract
Alzheimer’s disease (AD) is characterized by the accumulation of amyloid-β (Aβ). The genes that govern this process, however, have remained elusive. To this end, we combined distinct mouse strains with transcriptomics to directly identify disease-relevant genes. We show that AD model mice (APP-Tg) with DBA/2 genetic backgrounds have significantly lower levels of Aβ accumulation compared with SJL and C57BL/6 mice. We then applied brain transcriptomics to reveal the genes in DBA/2 that suppress Aβ accumulation. To avoid detecting secondarily affected genes by Aβ, we used non-Tg mice in the absence of Aβ pathology and selected candidate genes differently expressed in DBA/2 mice. Additional transcriptome analysis of APP-Tg mice with mixed genetic backgrounds revealed kinesin light chain-1 (Klc1) as an Aβ modifier, indicating a role for intracellular trafficking in Aβ accumulation. Aβ levels correlated with the expression levels of Klc1 splice variant E and the genotype of Klc1 in these APP-Tg mice. In humans, the expression levels of KLC1 variant E in brain and lymphocyte were significantly higher in AD patients compared with unaffected individuals. Finally, functional analysis using neuroblastoma cells showed that overexpression or knockdown of KLC1 variant E increases or decreases the production of Aβ, respectively. The identification of KLC1 variant E suggests that the dysfunction of intracellular trafficking is a causative factor of Aβ pathology. This unique combination of distinct mouse strains and model mice with transcriptomics is expected to be useful for the study of genetic mechanisms of other complex diseases.
To learn more about this research, please read the full research report entitled " Transcriptome analysis of distinct mouse strains reveals kinesin light chain-1 splicing as an amyloid-β accumulation modifier " at this page of the PNAS website.
Related link :