
Highly accurate calculation method for electronic configurations of magnetic molecules will lead to the development of molecular spintronics devices
When calculating electronic configurations in d and f orbitals which determine the properties of magnetic molecules with conventional first principles calculation methods, unreliable results are often obtained. Thus, the development of a first principles calculation method for predicting properties of magnetism in molecules with a high degree of accuracy has been sought. This time, the group established an important theoretical method for predicting new magnetic molecular devices toward the development of molecular spintronics devices. It is anticipated that this method will lead to research and development of practical magnetic molecular devices such as non-volatile molecular memory using single molecule magnet and quantum computers etc.
Spintronics, a technology to utilize electrons’ “charge” and “spin” degrees of freedom to make devices, has been actively studied in recent years and is attracting attention as a technology to bring high-speed and low power consumption devices. Above all, organometallic molecular devices can play a specific function at a single molecular level, so they can be further miniaturized. In this way, much interest in molecular spintronics has rapidly increased because it allows fabrication of the ultimate magnetic materials.
In order to design and develop high-performance devices using molecular spintronics, it is essential to understand the electronic configurations, which dominate properties of magnetism in the molecules, so theoretical approaches using first principles calculation are strongly desired.
In the first principles calculation approaches for analyzing the electronic configurations, density functional theory (DFT) is not applicable in some cases due to a complexity of the orbital degeneracy, changes in presence of the ligand field of molecules, and electronic correlation effects. Actually, even in ground-state electronic configurations, experimental observations and theoretical predictions are not consistent in many cases. In order to perform theoretical design for magnetic molecular materials with a high accuracy, it is necessary to overcome these difficulties.
The joint group of researchers led by Professor Tamio OGUCHI at Osaka University, Associate Professor Kohji NAKAMURA and Graduate Student Kenji NAWA at Mie University, and Distinguished Professor Michael Weinert at the University of Wisconsin-Milwaukee has developed a new first principles calculation approach that is able to analyze the electronic configurations with a high accuracy and explore the ground state by calculating the total energy of every conceivable electronic configuration (Figure 1 (b)) with the full-potential linearized augmented plane-wave (FLAPW) method that this group had developed on their own. Furthermore, this group quantitatively obtained a degree of correlation effects, effective on-site Coulomb interaction parameters, by devising a way of handling correlation effects between electrons, for using DFT+U method that considers many-body effects, although the values of these parameters are often qualitatively and empirically given that may lead to an inaccurate prediction. This group applied their method to metallocene molecues consisting of different transition-metal (M=V, Cr, Mn, Fe, Co, and Ni) atoms (Figure 1 (a)) to theoretically clarify the ground state and how the electronic configurations were built up. This results in an establishment of a world first, which will open an important avenue toward a new calculation method for predicting various properties of magnetic molecular materials.

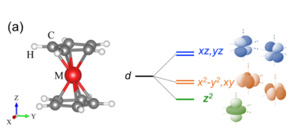
Fig 1. (a) Structure of an isolated metallocene where large (red), middle (gray), and small (white) circles indicate transition-metal (M), carbon, and hydrogen atoms, respectively, and schematic of the energy diagram of the crystal field splitting of transition-metal d orbitals in D5d symmetry. (b) Various d-orbital electronic configurations, in Mn-d5 system as an example, where black arrows indicate the occupied electrons. (Credit: Osaka University and Mie University)
To learn more about this research, please view the full research report entitled " Search for the ground-state electronic configurations of correlated organometallic metallocenes from constraint density functional theory " at this page of Physical Review B .
Related links